10-K: Annual report pursuant to Section 13 and 15(d)
Published on August 13, 2020
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
______________________________________________________________________________________________
FORM
______________________________________________________________________________________________
[
For the fiscal year ended
Commission file number:
______________________________________________________________________________________________
(Exact name of registrant as specified in its charter)
______________________________________________________________________________________________
(State or other jurisdiction of incorporation or organization)
(IRS Employer Identification No.)
(Address of principal executive offices)
(
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
|
|
|
|
|
Title of each class |
|
Trading Symbol(s) |
|
Name of each exchange on which registered |
|
|
Securities registered pursuant to Section 12(g) of the Act
None
______________________________________________________________________________________________
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes No ¨
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Act. Yes ¨ No
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes No ¨
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes No ¨
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company or an emerging growth company. See the definitions of “
|
|
|
|
Large Accelerated Filer |
x |
Accelerated Filer |
¨ |
Non-accelerated Filer |
¨ |
Smaller Reporting Company |
|
Emerging Growth Company |
|
|
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ¨
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No
The aggregate market value of the voting and non-voting common equity held by non-affiliates of registrant as of December 31, 2019 (the last business day of the registrant’s most recently completed second fiscal quarter), computed by reference to the closing sale price of such stock on the New York Stock Exchange, was $
At August 7, 2020, registrant had
Portions of the registrant’s definitive Proxy Statement to be delivered to stockholders in connection with the registrant’s 2020 Annual Meeting of Stockholders, to be filed subsequent to the date hereof, are incorporated by reference into Part III of this report.
TABLE OF CONTENTS
|
|
|
|
|
|
1 |
|
Part I |
Item 1 |
1 |
|
|
Item 1A |
18 |
|
|
Item 1B |
34 |
|
|
Item 2 |
34 |
|
|
Item 3 |
34 |
|
|
Item 4 |
34 |
|
Part II |
Item 5 |
35 |
|
|
Item 6 |
37 |
|
|
Item 7 |
Management’s Discussion and Analysis of Financial Condition and Results of Operations |
38 |
|
Item 7A |
Quantitative and Qualitative Disclosures About Market and Business Risks |
48 |
|
Item 8 |
50 |
|
|
Item 9 |
Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
82 |
|
Item 9A |
82 |
|
|
Item 9B |
86 |
|
Part III |
Item 10 |
87 |
|
|
Item 11 |
87 |
|
|
Item 12 |
Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
87 |
|
Item 13 |
Certain Relationships and Related Transactions, and Director Independence |
87 |
|
Item 14 |
87 |
|
Part IV |
Item 15 |
88 |
|
|
Item 16 |
89 |
|
|
|
90 |
As used in this 10-K, the terms “we”, “us”, “our” and “the Company” refer to ResMed Inc., a Delaware corporation, and its subsidiaries, on a consolidated basis, unless otherwise stated.
PART I
Cautionary Note Regarding Forward-Looking Statements
This report contains or may contain certain forward-looking statements and information that are based on the beliefs of our management as well as estimates and assumptions made by, and information currently available to, our management. All statements other than statements regarding historical facts are forward-looking statements. The words “believe,” “expect,” “intend,” “anticipate,” “will continue,” “will,” “estimate,” “plan,” “future” and other similar expressions, and negative statements of such expressions, generally identify forward-looking statements, including, in particular, statements regarding expectations of future revenue or earnings, expenses, new product development, new product launches, new markets for our products, litigation, and tax outlook. These forward-looking statements are made in accordance with the safe harbor provisions of the Private Securities Litigation Reform Act of 1995. You are cautioned not to place undue reliance on these forward-looking statements. Forward-looking statements reflect the views of our management at the time the statements are made and are subject to a number of risks, uncertainties, estimates and assumptions, including, without limitation, and in addition to those identified in the text surrounding such statements, those identified in Item 1A “Risk Factors” and elsewhere in this report.
In addition, important factors to consider in evaluating such forward-looking statements include changes or developments in healthcare reform, social, economic, market, legal or regulatory circumstances, including the impact of public health crises such as the novel strain of coronavirus (COVID-19) that has spread globally; changes in our business or growth strategy or an inability to execute our strategy due to changes in our industry or the economy generally, the emergence of new or growing competitors, the actions or omissions of third parties, including suppliers, customers, competitors and governmental authorities, and various other factors. If any one or more of these risks or uncertainties materialize, or underlying estimates or assumptions prove incorrect, actual results may vary significantly from those expressed in our forward-looking statements, and there can be no assurance that the forward-looking statements contained in this report will in fact occur.
ITEM 1 BUSINESS
General
We are a global leader in digital health and cloud-connected medical devices. We design innovative solutions to treat and keep people out of the hospital, empowering them to live healthier, higher-quality lives. Our digital health technologies and cloud-connected medical devices transform care for people with sleep apnea, chronic obstructive pulmonary disease, or COPD, and other chronic diseases. Our comprehensive out-of-hospital software platforms support the professionals and caregivers who help people stay healthy in the home or care setting of their choice. By enabling better care, our products improve quality of life, reduce the impact of chronic disease, and lower costs for consumers and healthcare systems.
Following our formation in 1989, we commercialized a treatment for obstructive sleep apnea, or OSA. This treatment, nasal continuous positive airway pressure, or CPAP, was the first successful noninvasive treatment for OSA. CPAP systems deliver pressurized air, typically through a nasal mask, to prevent collapse of the upper airway during sleep.
Since the development of CPAP, we have expanded our business by developing or acquiring a number of innovative products and solutions for a broad range of respiratory disorders including technologies to be applied in medical and consumer products, ventilation devices, diagnostic products, mask systems for use in the hospital and home, headgear and other accessories, dental devices, portable oxygen concentrators, or POCs, and cloud-based software informatics solutions to manage patient outcomes and customer and provider business processes. Today, we offer a comprehensive digital solution suite for patients with COPD, including those using inhalers or supplemental oxygen as well as non-invasive or invasive ventilation. We also provide management software to agencies providing out-of-hospital care, including home medical equipment, or HME, home health and hospice, skilled nursing, life plan community and senior living, and private duty services. Our growth has been fueled by geographic expansion, our research and product development efforts, acquisitions and an increasing awareness of sleep apnea and respiratory conditions like COPD as significant health concerns. We are also a leading provider of cloud-based software health applications and devices designed to provide connected care, improving patient outcomes and efficiencies for healthcare providers. These tools are designed to enable clinicians to manage more patients efficiently and effectively, as well as enable and encourage patients’ long-term adherence to and satisfaction with their therapy.
We employ approximately 7,800 people and sell our products in approximately 140 countries through a combination of wholly owned subsidiaries and independent distributors.
Our web site address is www.resmed.com. Information contained on our website is not part of or incorporated into this report. We make our periodic reports, together with any amendments, available on our website, free of charge, as soon as reasonably practicable after we electronically file or furnish the reports with the Securities and Exchange Commission, or SEC. The SEC maintains an internet site, www.sec.gov, which contains reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC.
Corporate History
ResMed Inc., a Delaware corporation, was formed in March 1994 as the ultimate holding company for our operating subsidiaries. In June 1995, we completed an initial public offering of common stock and our common stock began trading on the NASDAQ National Market. In September 1999, we transferred our principal listing to the New York Stock Exchange, or NYSE, trading under the ticker symbol “RMD”. In November 1999, we established a secondary listing of our common stock via Chess Depositary Instruments, or CDIs, on the Australian Stock Exchange (now known as the Australian Securities Exchange), or ASX, also under the symbol “RMD”. Ten CDIs on the ASX represent one share of our common stock on the NYSE.
Our Australian subsidiary, ResMed Holdings Limited, was originally organized in 1989 by Dr. Peter Farrell to acquire from Baxter Center for Medical Research Pty Limited, or Baxter, the rights to certain technology relating to CPAP treatment as well as Baxter’s existing CPAP device business. Baxter acquired the rights to the technology in 1987, and sold CPAP devices in Australia from 1988 until our acquisition of the business.
Since formation we have acquired a number of businesses, including distributors, suppliers, developers of medical equipment and related technologies and software solution providers.
Segment Information
We operate in two segments, which are the Sleep and Respiratory Care segment and the software as a service, or SaaS, segment. See Note 15 – Segment Information of the Notes to Financial Statements (Part II, Item 8) for financial information regarding segment reporting. Financial information about our revenues from and assets located in foreign countries is also included in the notes to our consolidated financial statements.
The Market
We are focused on the sleep and related respiratory care markets, both of which we believe are globally underpenetrated markets, and where we believe our products can improve patient outcomes, create efficiencies for our customers, help physicians and providers better manage chronic disease and reduce overall healthcare system costs. Additionally, our software solutions are focused on the out-of-hospital care market, which we believe is fragmented and underserved and where we see significant opportunity to transform and significantly improve out-of-hospital healthcare through a strategy of enabling better patient care, improving clinical decision support, and driving interoperability across out-of-hospital care settings.
Sleep and Respiratory Care
Sleep
Sleep is a complex neurological process that includes two distinct states: rapid eye movement, or REM, sleep and non-rapid eye movement, or non-REM, sleep. REM sleep, which is about 20-25% of total sleep experienced by adults, is characterized by a high level of brain activity, bursts of rapid eye movement, increased heart and respiration rates, and paralysis of many muscles. Non-REM sleep is subdivided into four stages that generally parallel sleep depth; stage 1 is the lightest and stage 4 is the deepest.
The upper airway has no rigid support and is held open by active contraction of upper airway muscles. Normally, during REM sleep and deeper levels of non-REM sleep, upper airway muscles relax and the airway narrows. Individuals with narrow upper airways or poor muscle tone are prone to temporary collapses of the upper airway during sleep, called apneas, and to near closures of the upper airway called hypopneas. These breathing events result in a lowering of blood oxygen concentration, causing the central nervous system to react to the lack of oxygen or increased carbon dioxide and signaling the body to respond. Typically, the individual subconsciously arouses from sleep, causing the throat muscles to contract, opening the airway. After a few gasping breaths, blood oxygen levels increase and the individual can resume a deeper sleep until the cycle repeats itself. Sufferers of OSA typically experience ten or more such cycles per hour. While these awakenings greatly impair the quality of sleep, the individual is not normally aware of these disruptions. In addition, OSA has been recognized as a cause of hypertension and a significant comorbidity for heart disease, stroke and diabetes.
A long-term epidemiology study published in 2013 estimated that 26% of adults age 30-70 have some form of obstructive sleep apnea. Another study published in 2019 estimated that mild to severe OSA impacts more than 936 million people worldwide, including 54 million Americans. Of those impacted, it was estimated that more than 424 million would have moderate to severe sleep apnea. Despite the high prevalence of OSA, there is a general lack of awareness of OSA among both the medical community and the general public. It is estimated that less than 20% of those with OSA have been diagnosed or treated. Many healthcare professionals are often unable to diagnose OSA because they are unaware that such non-specific symptoms as excessive daytime sleepiness, snoring, hypertension and irritability are characteristic of OSA.
While sleep apnea has been diagnosed in a broad cross-section of the population, until recently, it has typically been diagnosed among middle-aged men who are obese. However, we believe the importance of sleep apnea in women is increasingly being recognized, with nearly 40% of new PAP patients being female. A strong association has been discovered between sleep apnea and a number of cardiovascular and metabolic diseases. Studies have shown that sleep apnea is present in approximately 83% of patients with drug-resistant hypertension, approximately 77% of patients with obesity, approximately 76% of patients with chronic heart failure and approximately 72% of patients with type 2 diabetes.
Sleep-Disordered Breathing and Obstructive Sleep Apnea. Sleep-disordered breathing encompasses all disease processes that cause abnormal breathing patterns during sleep. Manifestations include OSA, central sleep apnea, or CSA, and hypoventilation syndromes that occur during sleep. Hypoventilation syndromes are generally associated with obesity, chronic obstructive lung disease and neuromuscular disease. OSA is the most common form of SDB.
Sleep fragmentation and the loss of the deeper levels of sleep caused by OSA can lead to excessive daytime sleepiness, reduced cognitive function, including memory loss and lack of concentration, depression and irritability. OSA sufferers also experience an increase in heart rate and an elevation of blood pressure during the cycle of apneas. Several studies indicate that the oxygen desaturation, increased heart rate and elevated blood pressure caused by OSA may be associated with increased risk of cardiovascular morbidity and mortality due to angina, stroke and heart attack. Patients with OSA have been shown to have impaired daytime performance in a variety of cognitive functions including problem solving, response speed and visual motor coordination, and studies have linked OSA to increased occurrences of traffic and workplace accidents.
Generally, an individual seeking treatment for the symptoms of OSA is referred by a general practitioner to a sleep specialist for further evaluation. The diagnosis of OSA typically requires monitoring the patient during sleep at either a sleep clinic or the patient’s home. During overnight testing, respiratory parameters and sleep patterns may be monitored, along with other vital signs such as heart rate and blood oxygen levels. Simpler tests, using devices such as our ApneaLink Air, or our automatic positive airway pressure devices, monitor airflow during sleep, and use computer programs to analyze airflow patterns. These tests allow sleep clinicians to detect any sleep disturbances such as apneas, hypopneas or subconscious awakenings.
Before 1981, the primary treatment for OSA was a tracheotomy, a surgical procedure to create a hole in the patient’s windpipe. Alternative surgical treatments have involved either uvulopalatopharyngoplasty, or UPPP, in which surgery is performed on the upper airway to remove excess tissue and to streamline the shape of the airway or implanting a device to add support to the soft palate. UPPP alone has a poor success rate; however, when performed in conjunction with multi-stage upper airway surgical procedures, a greater success rate has been claimed. These combined procedures, performed by highly specialized surgeons, are expensive and involve prolonged and often painful recovery periods. Surgical treatments are not considered first line therapy for OSA. Other alternative treatments available today include nasal surgery, mandibular advancement surgery, dental appliances, palatal implants, somnoplasty, nasal devices and electrical stimulation of the nerves or muscles. Alternative pharmaceutical therapy treatments are reported to be under development.
A variety of devices are marketed for the treatment of OSA. Most are only partially effective, but CPAP is a reliable treatment for all severities of OSA and is considered first-line therapy. Use of mandibular advancement devices is increasing as a second-line option in patients unable to use CPAP or those with mild OSA. These devices cause the mandible and tongue to be pulled forward and improve the dimensions of the upper airway. CPAP is a non-invasive means of treating OSA. CPAP was first used as a treatment for OSA in 1980 by Dr. Colin Sullivan, the past Chairman of our Medical Advisory Board and was commercialized for treatment of OSA in the United States in the mid-1980s. During CPAP treatment, a patient sleeps with a nasal interface connected to a small portable air device that delivers room air at a positive pressure. The patient breathes in air from the device and breathes out through an exhaust port in the interface. Continuous air pressure applied in this manner acts as a pneumatic splint to keep the upper airway open and unobstructed. Interfaces include nasal masks and nasal pillows. Sometimes, when a patient leaks air through their mouth, a full-face mask may need to be used, rather than a nasal interface.
CPAP is not a cure and, therefore, must be used on a nightly basis as long as treatment is required. Patient compliance has been a major factor in the efficacy of CPAP treatment. Early generations of CPAP units provided limited patient comfort and convenience. Patients experienced soreness from the repeated use of nasal masks and had difficulty falling asleep with the CPAP device operating at the prescribed pressure. In more recent years, product innovations to improve patient comfort and compliance have been developed. These include more comfortable patient interface systems; delay timers that gradually raise air pressure allowing the patient to fall asleep more easily; bilevel air devices, including our AirCurve 10 Series and Lumis devices, which provide different air pressures for inhalation and exhalation; heated humidification systems to make the airflow more comfortable; and autotitration devices that modulate the average pressure delivered during the night.
Respiratory Care
Our aim is to provide respiratory care solutions to patients with COPD, asthma, and other chronic respiratory diseases, such as overlap syndrome, obesity hypoventilation syndrome, or OHS, and neuromuscular disease, including amyotrophic lateral sclerosis, or ALS. We aim to improve their quality of life, slow down disease progression and reduce the costs of patient management.
Our products cover patients ranging from those who only require therapy from CPAP systems at night, to those who are dependent on non-invasive or invasive ventilation for life-support and those who require portable oxygen concentrators, or POCs. Our devices are predominantly used in the home and, to a lesser extent, in general hospital wards and respiratory wards. We supply CPAP and bilevel device systems, non-invasive and invasive ventilators, humidifiers and accessories, including masks and tubing. We also offer stationary and portable battery-powered oxygen concentrators for the administration of long-term oxygen therapy in the home as well as data management systems designed to improve the management of patients.
In March 2020, the World Health Organization declared the outbreak of a novel strain of coronavirus, or COVID-19, as a pandemic. We have observed increased demand for our ventilator devices and masks, and we are working with governments, health authorities, hospitals, physicians, and patients worldwide to assess their needs, and to deliver the ventilation therapy that is essential to treat the respiratory complications of COVID-19. Our primary focus is to maximize the availability of ResMed ventilators and other respiratory support devices for the patients that need them most.
Chronic Obstructive Pulmonary Disease. COPD encompasses a group of lung diseases defined by persistent airflow limitation, prolongation of exhalation and loss of elasticity in the lungs. It is a progressive and debilitating disease and is associated with an increased inflammatory response in the airways. Symptoms encountered with COPD include shortness of breath as well as chronic cough and increased sputum production. COPD includes diseases such as emphysema and chronic bronchitis. A recent study based on recent epidemiology data estimates that there are over 380 million people worldwide who suffer from COPD, the world’s third leading cause of death.
Patients with COPD can have different clinical presentations. Patients with chronic bronchitis present with low level of oxygen (hypoxemia) and elevated levels of carbon dioxide (hypercapnia), a chronic productive cough, cor pulmonale and are commonly overweight. Patients with emphysema have more normal blood gases, are usually thin and hyperinflated and have a decreased diffusion capacity. During sleep, chronic bronchitic patients display more severe hypoxemia. In general, the more hypoxic a COPD patient is during the day the more severe the hypoxemia experienced during sleep. Hypercapnia as a consequence of hypoventilation also occurs in COPD patients and is more pronounced in REM sleep. Some COPD patients may also suffer from comorbid OSA, a condition known as Overlap Syndrome.
Home non-invasive ventilation has the potential to reduce healthcare costs associated with the management of patients with severe COPD by significantly increasing the time between hospital readmissions.
Overlap Syndrome. In patients with Overlap Syndrome, CPAP has been shown to provide benefits in relation to reducing mortality, decreasing hospitalizations and improving lung function and gas exchange. Non-invasive ventilation, or NIV, has been demonstrated to improve outcomes in patients with acute exacerbations of COPD through its ability to improve respiratory acidosis and decrease dyspnea and work of breathing. It may also increase survival rates and reduce length of hospital stays, as well as reducing complicating factors such as ventilator-associated pneumonia. In patients with stable COPD, the advantages of home NIV are less clear, but clinical studies have shown improvements in dyspnea scores and health-related quality-of-life measures and reductions in hospital readmissions and intensive care stays.
Long-term oxygen therapy, or LTOT, is indicated in chronic respiratory failure patients. The administration of LTOT has been shown to increase survival rates in patients with severe resting hypoxemia. In hypoxemic COPD patients, LTOT is associated with a lower mortality compared to nocturnal oxygen therapy alone and also associated with improved health-related quality of life measures. In long-term COPD survivors with a history of congestive heart failure, LTOT is associated with a slowing of respiratory failure progression.
Obesity Hypoventilation Syndrome. OHS is characterized by the combination of obesity, chronic alveolar hypoventilation leading to daytime hypercapnia and hypoxia and sleep apnea after the exclusion of other causes of alveolar hypoventilation. An estimated 90% of patients with OHS also have OSA. In patients with OHS, positive airway therapy, both CPAP and NIV, has been shown to effectively treat upper airway obstruction and reverse daytime respiratory failure as well as reduce the work of breathing and improve respiratory drive.
Neuromuscular Disease. Neuromuscular disease is a broad term that encompasses many diseases that either directly (via intrinsic muscle pathology) or indirectly (via nerve pathology) impair the functioning of muscles. Symptoms of neuromuscular disease and respiratory failure include increasing generalized weakness and fatigue, dysphagia, dyspnoea on exertion and at rest, sleepiness, morning headache, difficulties with concentration and mood changes. Most neuromuscular diseases are characterized by progressive muscular impairment leading to loss of ambulation, being wheelchair-bound, swallowing difficulties, respiratory muscle weakness and, eventually, death from respiratory failure. Neuromuscular disorders can progress rapidly or slowly. Rapidly progressive conditions, such as ALS and Duchenne muscular dystrophy in teenagers, are characterized by muscle impairment which worsens over months and can result in death within a few years. Variable or slowly progressive conditions, such as myotonic muscular dystrophy, are characterized by muscle impairment that worsens over years and may mildly reduce life expectancy.
NIV treatment to patients with neuromuscular disease may lead to improvements in respiratory failure symptoms and daytime arterial blood gases. In ALS patients, NIV treatment has been associated with an improvement in quality of life measures, sleep-related symptoms and survival. Studies have demonstrated that patients with Duchenne muscular dystrophy may improve in quality of life measures and may increase chance of survival with NIV treatment.
Software as a Service
Due to multiple acquisitions, including Brightree in April 2016, HEALTHCAREfirst in July 2018 and MatrixCare in November 2018, our operations now include platforms that comprise our SaaS business. Our SaaS strategy is to develop a portfolio that assists durable medical equipment, or DME, HME, and other long-term care providers operate more effectively and efficiently across various out-of-hospital care settings. Our SaaS portfolio provides services across the HME, home health and hospice, skilled nursing, life plan community and senior living, and private duty services. Our offerings can help providers perform analytics, manage documentation and implement new reimbursement requirements as well as more effectively transfer data as patients move between different care settings.
Business Strategy
We believe that the sleep apnea and respiratory care markets will continue to grow in the future due to a number of factors, including increasing awareness of OSA, CSA and COPD, improved understanding of the role of sleep apnea treatment in the management of cardiac, neurologic, metabolic and related disorders, improved understanding of the role of non-invasive ventilation in the management of COPD, and an increase in the use of digital and product technology to improve patient outcomes and create efficiencies for customers and providers. Additionally, the continued impact of COVID-19 or a resurgence of COVID-19 may create more demand for our ventilator products. Our strategy for expanding our business operations and capitalizing on the growth of the sleep apnea and respiratory care markets, as well as growth in out-of-hospital care settings, consists of the following key elements:
Continue Product Development and Innovation in Sleep Apnea Products. We are committed to ongoing innovation in developing products for the diagnosis and treatment of sleep apnea. We have been a leading innovator of products designed to treat sleep apnea more effectively, increase patient comfort and encourage compliance with prescribed therapy. In recent years we have introduced a full suite of masks in our AirTouch and AirFit ranges, advanced and expanded the integrations of our therapy-based software solutions, including AirView, to promote greater patient adherence and during the COVID-19 pandemic, we released ResMed MaskSelector in the United States, an easy-to-use digital tool to make mask selection and sizing easier and more effective, both remotely and during in-person clinical setups. We believe that the combination of continued product development, product and technology acquisitions and innovation are key factors to our ongoing success. Our recent acquisitions have included a portfolio of sleep apnea products through our acquisition of Curative Medical. Approximately 16% of our employees are devoted to research and development activities.
Continue Product Development and Innovation in Respiratory Care Products. We are committed to ongoing innovation of our respiratory care products that serve the needs of patients with COPD and neuromuscular diseases. With the addition of Inova Labs POCs and our non-invasive ventilator devices with masks and accessories, we intend to continue to expand and enhance our product offerings in this area. In recent years, we launched Mobi, which is our first ResMed-branded portable oxygen concentrator as well as advanced and expanded the integrations of our therapy-based software solutions including AirView for Respiratory Care, enabling clinicians to remotely monitor patients on some ventilation devices and bilevel devices. Additionally, we acquired a digital health platform for inhalers through our acquisition of Propeller Health in 2019, rounding out our portfolio to treat COPD patients through their therapy journey across different stages of their disease.
Expand SaaS Solutions in Out-of-Hospital Care Settings. Our vision is to transform and significantly improve out-of-hospital (OOH) healthcare through a strategy of enabling better patient care, improving clinical decision support, and driving interoperability across out-of-hospital healthcare settings. Since acquiring Brightree in 2016, plus MatrixCare and HEALTHCAREfirst in 2018, we offer software solutions across multiple out-of-hospital healthcare settings including HME, home health and hospice, skilled nursing, life plan communities, senior living and private duty. We are connecting capabilities across the platforms in these out-of-hospital care settings to help our customers be more efficient, better serve people, keep them out-of-hospital, and in lower-cost, higher-quality care settings. Today, our SaaS solutions serve OOH customers combining over 90 million individual accounts.
Expand Geographic Presence. We market our products in more than 140 countries to sleep clinics, home healthcare dealers, patients and third-party payors. We intend to increase our sales and marketing efforts in our principal markets, as well as expand the depth of our presence in other high-growth geographic regions. In 2016, we acquired Curative Medical to invest in the China market and expand our growth potential in sleep apnea, COPD and respiratory care in China. In 2019, we acquired HB Healthcare, a privately owned HME that serves both reimbursed and cash-pay customers of sleep and respiratory care devices in South Korea.
Increase Public and Clinical Awareness. We continue to expand our existing promotional activities to increase awareness of sleep apnea, COPD and other clinical conditions that can be treated with our industry-leading solutions. These promotional activities target both the population predisposed to sleep apnea and medical specialists, such as pulmonologists, sleep medicine specialists, primary care physicians, cardiologists, neurologists and other medical subspecialists who treat these conditions and their associated comorbidities. We target special interest groups, including the National Stroke Association, the American Heart Association, COPD Foundation and the National Sleep Foundation, to further increase awareness of the relationship between OSA, COPD, neuromuscular disease and comorbidities such as cardiac disease, diabetes, hypertension and obesity. The programs also support our efforts to inform the community of the dangers of sleep apnea with regard to occupational health and safety, especially in the transport industry. We have helped establish a center for clinical care and medical research at the University of California, San Diego in the fields of sleep apnea and COPD.
Expand into New Clinical Applications. We continually seek to identify new applications of our technology for significant unmet medical needs. Studies have established a clinical association between OSA and both stroke and congestive heart failure, and have recognized sleep apnea as a cause of hypertension or high blood pressure. Research also indicates that sleep apnea is independently associated with glucose intolerance and insulin resistance. Additionally, research supported by ResMed has demonstrated that the addition of non-invasive ventilation to patients with severe COPD who are receiving oxygen therapy, provides meaningful clinical benefits to the patient, and the broader healthcare system. We maintain close working relationships with a number of prominent physicians to explore new medical applications for our products and technology.
Leverage the Experience of our Management Team. Our senior management team has extensive experience in the medical device industry in general, and in the fields of sleep apnea, respiratory care and healthcare informatics in particular. We intend to continue to leverage the experience and expertise of these individuals to maintain our innovative approach to the development of products and solutions, and to increase awareness of the serious medical problems caused by sleep apnea and the use of oxygen, non-invasive ventilation, and in-home life support ventilation to treat COPD.
Products
Our portfolio of products includes devices, diagnostic products, mask systems, headgear and other accessories, dental devices, POCs and cloud-based software informatics solutions. For purposes of the following discussion, we refer to our air flow generators, ventilators and oxygen concentrators collectively as devices.
Devices
We produce cloud-connected CPAP, APAP, bilevel, and ASV devices that deliver positive airway pressure through a patient interface, either a mask or cannula. Our APAP devices, known as AutoSet, are based on a proprietary technology to monitor breathing and can also be used in the diagnosis, treatment and management of OSA. During fiscal year 2017, we launched AirMini, a small portable CPAP combining the same proven therapy modes used in the AirSense 10 with effective waterless humidification enabling portable convenience. We also acquired a line of Chinese-developed and manufactured sleep and ventilation devices with the acquisition of Curative Medical in fiscal year 2016. Devices in total accounted for approximately 51%, 52% and 56% of our net revenues in fiscal years 2020, 2019 and 2018, respectively.
The tables below provide a selection of products, as known by our trademarks.
CPAP |
DESCRIPTION |
AirSense 10 Elite |
An advanced fixed-pressure therapy device with an integrated humidifier and built-in wireless connectivity. It is designed to be intuitive and easy-to-use.
|
AirSense 10 CPAP |
The AirSense 10 CPAP is a fixed-pressure therapy device and built-in wireless connectivity. It also provides compliance, AHI and leak data reporting.
|
AUTOSET |
DESCRIPTION |
AirSense 10 Auto |
A premium auto-adjusting therapy device featuring AutoRamp™ with sleep onset detection, expiratory pressure relief (EPR™) and Easy-Breathe technology. The device also features built-in wireless connectivity.
|
AirMini |
A small portable CPAP device featuring the same auto-adjusting therapy modes used in the AirSense™ 10 Auto. The device also features built-in Bluetooth connectivity and effective waterless humidification enabled by HumidX technology.
|
BILEVEL |
DESCRIPTION |
AirCurve 10 S |
A bilevel device for patients who need extra pressure support or find it difficult to adjust to therapy on a fixed pressure continuous positive airway pressure device. The device features built-in wireless connectivity and works with our AirView™ patient monitoring software.
|
AirCurve 10 V Auto |
An auto-adjusting bilevel device for patients who need greater pressure support to treat their obstructive sleep apnea. The device features built-in wireless connectivity and works with our AirView™ patient monitoring software.
|
AirCurve 10 ST |
A bilevel device with backup rate that provides exceptional patient-ventilator synchrony, reducing the work of breathing so patients remain comfortable and well ventilated. The device features built-in wireless connectivity and works with our AirView™ patient monitoring software.
|
AirCurve 10 ASV and CS |
Adaptive servo-ventilators specifically designed to treat patients exhibiting central sleep apnea (CSA), mixed sleep apnea and periodic breathing, with or without obstructive sleep apnea. These devices also feature built-in wireless connectivity and works with our AirView™ patient monitoring software.
|
VENTILATION |
DESCRIPTION |
Stellar 100 and 150 |
Pressure support and volume non-invasive ventilators with invasive capabilities designed to suit a range of environments and for various respiratory patient types.
|
Astral 100 and 150 |
Pressure support and volume ventilators for invasive and non-invasive purposes so it can be used from the hospital to the home.
|
Lumis 100 and 150 |
Pressure support non-invasive ventilators that support a variety of therapy modes with built-in wireless connectivity and integrated humidification.
|
Lumis ST-A |
A Pressure support non-invasive ventilator that supports a variety of therapy modes with built-in wireless connectivity, integrated humidification and a range of fixed and adjustable alarms.
|
Mobi |
A portable oxygen concentrator system with a lightweight design and extended battery life to promote greater mobility for patients on oxygen therapy.
|
Mask Systems, Diagnostic Products, Accessories and Other Products
Masks, diagnostic products and accessories together accounted for approximately 37%, 37% and 38% of our net revenues in fiscal years 2020, 2019 and 2018, respectively.
Mask Systems
Mask systems are one of the most important elements of sleep apnea treatment systems. Masks are a primary determinant of patient comfort and as such may drive or impede patient compliance with therapy. We have been a consistent innovator in small nasal, nasal pillows and full-face masks, by improving patient comfort while minimizing size and weight.
The table below provides an of overview of our mask systems by category.
CATEGORY |
DESCRIPTION |
Minimalist |
AirFit F30, AirFit P10 and AirFit N30 minimalist masks feature our lightest, lowest profile designs. The features of these masks are focused on minimizing contact with the patient’s face to reduce red marks and irritation.
|
Freedom |
AirFit N30i, AirFit P30i and AirFit F30i freedom masks, which feature top-of-head tubing design allowing flexibility to easily switch sleep positions.
|
Ultra Soft |
The AirTouch F20 mask features a soft and breathable AirTouch cushion designed to enhance CPAP mask comfort.
|
Universal Fit |
AirFit F20 and AirFit N20 masks are designed to fit a wide range of faces due to the InfinitySeal silicone cushion that adapts to unique facial contours, which increases comfort, improves the fit and reduces leakage.
|
Diagnostic Products
We market sleep recorders for the diagnosis and titration of sleep apnea in sleep clinics and hospitals. These diagnostic systems record relevant respiratory and sleep data, which can be analyzed by a sleep specialist or physician who can then tailor an appropriate OSA treatment regimen for the patient.
PRODUCTS |
DESCRIPTION |
ApneaLink Air |
A portable diagnostic device which measures oximetry, respiratory effort, pulse, nasal flow and snoring. Works with AirView Diagnostics to provide comprehensive diagnostic solution to clinicians.
|
Connected Solutions and Other Products
We have a suite of products that are designed to allow fewer professionals to manage more patients and empower patients to track their own health outcomes. We are expanding our cloud-based patient management and engagement platforms, such as AirView, enabling remote monitoring, over-the-air trouble shooting and changing of device settings, U-Sleep enabling automated patient coaching through a text, email or interactive voice phone call and myAir, a patient engagement application that provides sleep data and a daily score based on their previous night’s data.
PRODUCTS |
DESCRIPTION |
AirView |
A cloud-based system enabling remote monitoring and changing of patients’ device settings. AirView also makes it easier to simplify workflows and collaborate more efficiently across the patient’s care network.
|
myAir |
A personalized therapy management application for patients with sleep-disordered breathing providing support, education and troubleshooting tools for increased patient engagement and improved compliance.
|
U-Sleep |
A compliance monitoring solution that enables HMEs to streamline their sleep programs to achieve better business and patient outcomes.
|
Connectivity Module |
A module providing cellular connection between our compatible ventilation devices (i.e. Astral Stellar) and our AirView™ system.
|
|
Propeller Solutions |
Propeller's inhaler sensors track medication usage and pair with a companion smartphone application, giving people with asthma or COPD a better understanding of their disease and while promoting increased adherence to treatment. The Propeller Provider Portal gives clinicians the timely and accurate information they need to make better treatment decisions.
|
SaaS Products
Following multiple acquisitions, including Brightree in April 2016, HEALTHCAREfirst in July 2018 and MatrixCare in November 2018, we now supply out-of-hospital software products designed to support the professionals and caregivers helping people stay healthy in the home or care setting of their choice. SaaS revenue accounted for approximately 12%, 11% and 7% of our net revenue in fiscal years 2020, 2019 and 2018, respectively.
PRODUCTS |
DESCRIPTION |
Brightree solutions |
Brightree enables out-of-hospital care organizations to improve their business performance and deliver better health outcomes. As an industry-leading cloud-based healthcare IT company, Brightree provides solutions and services for thousands of organizations in home medical equipment and pharmacy, orthotic and prosthetic, and home infusion.
|
|
HEALTHCAREfirst solutions
|
HEALTHCAREfirst offers electronic health record, or EHR, software, billing and coding services, and advanced analytics that enable home health and hospice agencies to optimize their clinical, financial and administrative processes.
|
MatrixCare solutions |
MatrixCare’s EHR software as a service solutions are used by skilled nursing and senior living providers, life plan communities (CCRCs), and home health and hospice organizations to prosper in an ever-changing healthcare system.
|
Product Development and Clinical Trials
We have a strong track record of innovation in the sleep and respiratory care markets. In 1989, we introduced our first CPAP device. Since then we have been committed to an ongoing program of product advancement and development. Currently, our product development and clinical trial efforts are focused on not only improving our current product offerings and usability, but also expanding into new product applications.
We continually seek to identify new applications of our technology for significant unmet medical needs. Sleep apnea is associated with a number of symptoms beyond excessive daytime sleepiness and irritability. Studies have established a clinical association between untreated sleep apnea and systemic hypertension, diabetes, coronary artery disease, stroke, atrial fibrillation, congestive heart failure, and mortality.
Across the sleep and respiratory care platforms, we support clinical trials in many countries including the United States, Germany, Netherlands, France, Japan, the United Kingdom, Switzerland, China, Spain, Canada, Singapore and Australia to develop new clinical applications for our technology. We have also begun presenting and publishing research findings based on the industry-leading connectivity platform and data assets that are unique to us. We continue to support some of the largest sleep apnea studies in history by performing advanced statistical analyses on millions of clinical data points using real-world data.
We consult with physicians at major medical centers throughout the world to identify clinical and technological trends in the treatment of sleep apnea, COPD and the other conditions associated with these diseases. New product ideas are also identified by our marketing staff, direct sales force and network of distributors, customers, clinicians and patients.
Sales and Marketing
We currently market our products in more than 140 countries through a network of distributors and our direct sales force. We attempt to tailor our marketing approach to each national market, based on regional awareness of sleep apnea as a health problem, physician referral patterns, consumer preferences and local reimbursement policies. See Note 15 – Segment Information of the Notes to Consolidated Financial Statements (Part II, Item 8) for financial information about our geographic areas.
United States, Canada and Latin America. Our products are typically purchased by a home healthcare dealer who then sells the products to the patient. The decision to purchase our products, as opposed to those of our competitors, is made or influenced by one or more of the following individuals or organizations: the prescribing physician and his or her staff; the home healthcare dealer; the insurer and the patient. In the United States, Canada and Latin America, our sales and marketing activities are conducted through a field sales organization made up of regional territory representatives, program development specialists and regional sales directors. Our field sales organization markets and sells products to home healthcare dealer branch locations throughout the United States, Canada and Latin America.
We also market our products directly to physicians and sleep clinics. Patients who are diagnosed with OSA or another respiratory condition and prescribed our products are typically referred by the diagnosing physician or sleep clinic to a home healthcare dealer to fill the prescription. The home healthcare dealer, in consultation with the referring physician, will assist the patient in selecting the equipment, fit the patient with the appropriate mask and set the device pressure to the prescribed level.
Our SaaS solutions are sold to providers of healthcare in various out-of-hospital settings. We market and sell our Brightree business management software and service solutions to providers in the U.S. and our primary markets are HME, pharmacy, home infusion, orthotics and prosthetics. Our sales activities for Brightree products are conducted through a sales organization made up of strategic account managers, sales engineers and sales directors. We develop, market and sell our MatrixCare care management and related ancillary solutions to providers in the U.S. and our primary markets are senior living, skilled nursing; life plan communities; home health, home care, and hospice agencies as well as related accountable care organizations. Our MatrixCare management solutions are primarily sold through direct sales and ancillary solutions are sold both through direct sales and channel partners.
Combined Europe, Asia and other markets. We market our products in most major countries in combined Europe, Asia and other markets. We have wholly-owned subsidiaries in Austria, Czech Republic, Denmark, Finland, France, Germany, Ireland, Netherlands, Norway, Poland, Sweden, Switzerland, the United Kingdom, Australia, China, India, Japan, Korea, New Zealand, Taiwan, and Thailand. We use a combination of our direct sales force and independent distributors to sell our products in combined Europe, Asia and other markets. We select independent distributors in each country based on their knowledge of respiratory medicine and a commitment to sleep apnea therapy. In countries where we sell our products direct, a local senior manager is responsible for direct national sales. In many countries we sell our products to home healthcare dealers or hospitals who then sell the products to the patients. In Germany, Australia, New Zealand, and South Korea, we also operate a home healthcare company, in which we provide products and services directly to patients.
We do not sell our SaaS products in combined Europe, Asia and other markets.
Manufacturing
We operate a globally distributed manufacturing network designed for supply chain resilience, that is intended to control costs and minimize risks. Our manufacturing operations consist of specialist component production as well as assembly and testing of our devices, masks and accessories. Of the numerous raw materials, parts and components purchased for assembly of our therapeutic and diagnostic sleep disorder products, many are off-the-shelf items available from multiple vendors. We also purchase uniquely configured components from various suppliers, including some who are single-source suppliers for us. Any reduction or halt in supply from one of these single-source suppliers could limit our ability to manufacture our products or devices until a replacement supplier is found and qualified. We generally manufacture to our internal sales forecasts and fill orders as received. We strive for continuous improvement in manufacturing processes to deliver year-on-year improvement in output, cost and product quality. Each manufacturing site and team are responsible for the quality of their product group and decisions are based on performance and quality measures, including customer feedback.
Our quality management system is based upon the requirements of ISO 9001, ISO 13485, FDA Quality System Regulations for Medical Devices, the Medical Device Directive (93/42/EEC) and other applicable regulations for the markets in which we sell. Our main manufacturing sites are certified to ISO 13485 and audited at regular intervals by a Notified Body. Additionally, our Sydney, Loyang, San Diego, Atlanta and Moreno Valley sites are certified under the Medical Device Single Audit Program or MDSAP, an audit of medical device manufacturers’ quality management system to satisfy multiple regulatory requirements. MDSAP audits are conducted by a MDSAP recognized auditing organization and can fulfill the needs of multiple regulatory jurisdictions (i.e. Australia, Brazil, Canada, Japan, and the United States of America).
Our main manufacturing facilities are located in Loyang, Singapore; Sydney, Australia; Chatsworth, California; Johor Bahru, Malaysia; Atlanta, Georgia and Suzhou, China. We are establishing a new manufacturing facility in Tuas, Singapore that will eventually replace our Loyang facility. Refer to Item 2 for additional details on these properties.
Third-Party Coverage and Reimbursement
The cost of medical care in many of the countries in which we operate is funded in substantial part by government and private insurance programs. In Germany and Korea, we receive payments directly from these payors but in other countries, although we do not generally receive payments for our products directly from these payors, our success in major markets depends on the ability of patients to obtain coverage and adequate reimbursement from third-party payors for our products.
In the United States, our products are purchased primarily by home healthcare dealers, hospitals or sleep clinics, who invoice third-party payors directly for reimbursement. Domestic third-party payors include government payors such as Medicare and Medicaid and commercial health insurance plans. These payors may deny coverage and reimbursement if they determine that a device is not used in accordance with certain covered treatment methods, or is experimental, unnecessary or inappropriate. The long-term trend towards cost-containment, through managed healthcare, or other legislative proposals to reform healthcare, could control or significantly influence the purchase of healthcare services and products and could result in lower prices for our products. In some foreign markets, such as France, Germany, and Japan, government reimbursement is currently available for purchase or rental of our products, subject to constraints such as price controls or unit sales limitations. In Australia, China, and some other foreign markets, there is currently limited or no reimbursement for devices that treat OSA.
Healthcare reform in the United States continues to bring significant changes to the third-party payor landscape. In 2011, the Centers for Medicare & Medicaid Services, or CMS, implemented the competitive bidding program, which included DME that we manufacture and develop, specifically, oxygen, CPAP and respiratory assist devices (or bilevel devices), and related supplies and accessories. CMS is required by law to recompete these contracts at least once every three years. In addition, the 2010 Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the ACA, required CMS to roll out the competitive bidding process nationally or adjust prices in non-competitive bidding areas, also known as the non-bid or Round 3 areas, to match competitive bidding prices by 2016. CMS phased in the new rates beginning January 1, 2016, and the rates became fully effective July 1, 2016. The implementation of the competitive acquisition program has resulted in reduced Medicare payment for oxygen, CPAP and respiratory assist devices, and related supplies and accessories in both competitive bidding areas and non-competitive bidding areas. Through an Interim Final Rule issued in May 2018, CMS increased the fee schedule amounts for certain DME in non-bid areas that qualify as rural and non-contiguous, setting payment for these areas for June 1, 2018 to December 31, 2018 at a 50/50 blended reimbursement rate based on the pre-competitive bidding reimbursement rate and the adjusted reimbursement rate set through competitive bidding.
Due to the lapse of competitive bid contracts as of December 31, 2018, effective January 1, 2019, Medicare beneficiaries may receive DME from any Medicare-enrolled supplier until new contracts are in effect under the next round of competitive bidding, which is expected to take effect on January 1, 2021. Pricing in competitive bidding areas and non-rural, contiguous non-bid areas will continue to use adjusted fee schedule amounts, subject to annual Consumer Price Index (CPI) adjustments, beginning in 2019, until the next bidding round takes place. CMS also extended the blended fee schedule amounts for non-bid rural and non-contiguous areas through December 31, 2020. Under the Coronavirus Aid, Relief, and Economic Security Act, or the CARES Act, the blended fee schedule amounts for non-bid rural and non-contiguous areas was extended through the end of the COVID-19 public health emergency, should it extend beyond December 31, 2020, and a blended fee schedule amount was implemented for all other areas for the same period.
In the next round of Durable Medical Equipment, Prosthetics, Orthotics and Supplies (DMEPOS) competitive bidding program, expected to take effect on January 1, 2021, there have been some revisions to the bidding methodology including the plan to implement surety bond requirements, lead item pricing, and setting reimbursement rates at the maximum winning bid rate instead of the median winning bid rate. Although CMS previously expanded the categories of devices subject to competitive bidding to include non-invasive ventilators, or NIVs, starting in 2021, in response to the COVID-19 pandemic, CMS removed NIVs from Round 2021 of the DMEPOS Competitive Bidding Program.
The ACA, which was passed both to expand the number of individuals with healthcare coverage and to develop additional revenue sources, also included, among other things, a deductible excise tax equal to 2.3% of the price for which medical devices are sold in the United States on any entity that manufactures or imports medical devices, with limited exceptions, beginning in 2013. However, this excise tax was subsequently suspended by the U.S. Congress for medical device sales, beginning in 2016 and permanently repealed, effective January 1, 2020. The ACA also provided for a number of Medicare regulatory requirements, including new face-to-face encounter requirements for DME and home health services.
We cannot predict at this time the full impact that the ACA, or any U.S. legislation enacted in the future, will have on our revenues, profit margins, profitability, operating cash flows and results of operations. There have been judicial and Congressional challenges to certain aspects of the ACA, as well as recent efforts by the Trump administration to repeal or replace certain aspects of the ACA, and we expect such challenges and amendments to continue. For example, the Tax Cuts and Jobs Act of 2017 includes a provision repealing, effective January 1, 2019, the tax-based shared responsibility payment imposed by the ACA on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate.” On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas ruled that the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as part of the U.S. Tax Act, the remaining provisions of the ACA are invalid as well. On December 18, 2019, the U.S. Court of Appeals for the 5th Circuit upheld the District Court ruling that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. On March 2, 2020, the U.S. Supreme Court granted the petitions for writs of certiorari to review this case, although it is unclear when or how the Supreme Court will rule. It is also unclear how other efforts to challenge, repeal or replace the ACA will impact the law or our business.
Service and Warranty
We generally offer either one-year or two-year limited warranties on our devices. In some regions and for certain customers we also offer extended warranties on our devices for one to three years in addition to our limited warranty. Warranties on mask systems are for 90 days. Our distributors either repair our products with parts supplied by us or arrange shipment of products to our facilities for repair or replacement. We receive returns of our products from the field for various reasons. We believe that the level of returns experienced to date is consistent with levels typically experienced by manufacturers of similar devices. We provide for warranties and returns based on historical data.
Competition
The markets for our products and services are highly competitive. We believe that the principal competitive factors in all of our markets are product features, value-added solutions, reliability and price. Customer support, reputation and efficient distribution are also important factors. We compete on a market-by-market basis with various companies, some of which have greater financial, research, manufacturing and marketing resources than us. The disparity between our resources and those of our competitors may increase as a result of the trend towards consolidation in the healthcare industry. In addition, some of our competitors are affiliates of customers of ours, which may make it difficult to compete with them.
Our primary Sleep and Respiratory Care competitors include Philips BV; Fisher & Paykel Healthcare Corporation Limited; DeVilbiss Healthcare; Apex Medical Corporation; BMC Medical Co. Ltd.; and regional manufacturers. Finally, our products compete with surgical procedures, nerve stimulation devices and dental appliances designed to treat OSA and other sleep apnea-related respiratory conditions. The development of new or innovative procedures or devices by others could result in our products becoming obsolete or noncompetitive, which would harm our revenues and financial condition.
For our SaaS business, the market is highly competitive, rapidly evolving, and subject to changing technology, low barriers to entry, shifting customer needs and frequent introductions of new products and services. The development of new or innovative solutions by others could result in our solutions becoming obsolete or noncompetitive, which would harm our revenues and financial condition.
Any product developed by us will have to compete for market acceptance and market share. An important factor in such competition may be the timing of market introduction of competitive products and solutions. Accordingly, the speed with which we can develop products and solutions, complete clinical testing and regulatory clearance processes and provide commercial supply of products and solutions to the market are important competitive factors. In addition, our ability to compete will continue to be dependent on successfully protecting our patents and other intellectual property.
Patents and Proprietary Rights and Related Litigation
We rely on a combination of patents, designs, trademarks, trade secrets, copyrights, and non-disclosure agreements to protect our proprietary technology and rights. Some of these patents, patent applications and designs relate to significant aspects and features of our products. We believe the combination of these rights, in aggregate, are of material importance to each of our businesses. Through our various subsidiaries, as of the date of this report, we own or have licensed rights to over 6,200 pending, allowed or granted patents and designs. Patents and designs have various statutory terms based on the legislation in individual jurisdictions which may be subject to change. Of our patents, 570 U.S. patents and 1,452 foreign patents are due to expire in the next five years. We believe that the expiration of these patents will not have a material adverse impact on our competitive position.
Litigation has been necessary in the past and may be necessary in the future to enforce patents issued to us, to protect our rights, or to defend third-party claims of infringement by us of the proprietary rights of others. The defense and prosecution of patent claims, including pending claims, as well as participation in other inter-party proceedings, can be expensive and time-consuming, even in those instances in which the outcome is favorable to us. Patent laws regarding the enforceability of patents vary from country to country. Therefore, there can be no assurance that patent issues will be uniformly resolved, or that local laws will provide us with consistent rights and benefits.
Government Regulations
FDA
Our products are subject to extensive regulation particularly as to safety, efficacy and adherence to FDA Quality System Regulation, and related manufacturing standards. Medical device products are subject to rigorous FDA and other governmental agency regulations in the United States and similar regulations of foreign agencies abroad. The FDA regulates the design, development, research, preclinical and clinical testing, introduction, manufacture, advertising, labeling, packaging, marketing, distribution, import and export, and record keeping for such products, in order to ensure that medical products distributed in the United States are safe and effective for their intended use. In addition, the FDA is authorized to establish special controls to provide reasonable assurance of the safety and effectiveness of most devices. Non-compliance with applicable requirements can result in import detentions, fines, civil and administrative penalties, injunctions, suspensions or losses of regulatory approvals, recall or seizure of products, operating restrictions, refusal of the government to approve product export applications or allow us to enter into supply contracts, and criminal prosecution.
Unless an exemption applies, the FDA requires that a manufacturer introducing a new medical device or a new indication for use of an existing medical device obtain either a Section 510(k) premarket notification clearance or a premarket approval, or PMA, before introducing it into the U.S. market. The type of marketing authorization is generally linked to the classification of the device. The FDA classifies medical devices into one of three classes (Class I, II or III) based on the degree of risk the FDA determines to be associated with a device and the level of regulatory control deemed necessary to ensure the device’s safety and effectiveness.
Our products currently marketed in the United States are marketed pursuant to 510(k) pre-marketing clearances and are either Class I or Class II devices. The process of obtaining a Section 510(k) clearance generally requires the submission of performance data and often clinical data, which in some cases can be extensive, to demonstrate that the device is “substantially equivalent” to a device that was on the market before 1976 or to a device that has been found by the FDA to be “substantially equivalent” to such a pre-1976 device, a predecessor device is referred to as “predicate device.” As a result, FDA clearance requirements may extend the development process for a considerable length of time. In addition, in some cases, the FDA may require additional review by an advisory panel, which can further lengthen the process. The PMA process, which is reserved for new devices that are not substantially equivalent to any predicate device and for high-risk devices or those that are used to support or sustain human life, may take several years and requires the submission of extensive performance and clinical information.
Medical devices can be marketed only for the indications for which they are cleared or approved. After a device has received 510(k) clearance for a specific intended use, any change or modification that significantly affects its safety or effectiveness, such as a significant change in the design, materials, method of manufacture or intended use, may require a new 510(k) clearance or PMA approval and payment of an FDA user fee. The determination as to whether or not a modification could significantly affect the device’s safety or effectiveness is initially left to the manufacturer using available FDA guidance; however, the FDA may review this determination to evaluate the regulatory status of the modified product at any time and may require the manufacturer to cease marketing and recall the modified device until 510(k) clearance or PMA approval is obtained. The manufacturer may also be subject to significant regulatory fines or penalties.
Any devices we manufacture and distribute pursuant to clearance or approval by the FDA are subject to pervasive and continuing regulation by the FDA and certain state agencies. These include product listing and establishment registration requirements, which help facilitate FDA inspections and other regulatory actions. As a medical device manufacturer, all of our manufacturing facilities are subject to inspection on a routine basis by the FDA. We are required to adhere to applicable regulations setting forth detailed cGMP requirements, as set forth in the QSR, which require, manufacturers, including third-party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all phases of the design and manufacturing process. Noncompliance with these standards can result in, among other things, fines, injunctions, civil penalties, recalls or seizures of products, total or partial suspension of production, refusal of the government to grant 510(k) clearance or PMA approval of devices, withdrawal of marketing approvals and criminal prosecutions. We believe that our design, manufacturing and quality control procedures are in compliance with the FDA’s regulatory requirements.
We must also comply with post-market surveillance regulations, including medical device reporting, or MDR, requirements which require that we review and report to the FDA any incident in which our products may have caused or contributed to a death or serious injury. We must also report any incident in which our product has malfunctioned if that malfunction would likely cause or contribute to a death or serious injury if it were to recur.
Labeling and promotional activities are subject to scrutiny by the FDA and, in certain circumstances, by the Federal Trade Commission. Medical devices approved or cleared by the FDA may not be promoted for unapproved or uncleared uses, otherwise known as “off-label” promotion. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including substantial monetary penalties and criminal prosecution.
Sales of medical devices outside the United States are subject to regulatory requirements that vary widely from country to country.
EEA
In the European Economic Area, (which is comprised of the 27 member states of the European Union plus Norway, Iceland and Liechtenstein), or EEA, manufacturers of medical devices need to comply with the Essential Requirements laid out in Annex I to the EU Medical Devices Directive (Council Directive 93/42/EEC). Compliance with these requirements is a prerequisite to be able to affix the CE mark to medical devices, without which they cannot be marketed or sold in the EEA. To demonstrate compliance with the Essential Requirements and obtain the right to affix the CE Mark, manufacturers of medical devices must undergo a conformity assessment procedure, which varies according to the type of medical device and its classification. Except for low-risk medical devices (Class I with no measuring function and which are not sterile), where the manufacturer can issue an EC Declaration of Conformity based on a self-assessment of the conformity of its products with the Essential Requirements, a conformity assessment procedure requires the intervention of a Notified Body, which is an organization designated by a competent authority of an EEA country to conduct conformity assessments. Depending on the relevant conformity assessment procedure, the Notified Body would audit and examine the Technical File and the quality system for the manufacture, design and final inspection of the devices. The Notified Body issues a CE Certificate of Conformity following successful completion of a conformity assessment procedure conducted in relation to the medical device and its manufacturer and their conformity with the Essential Requirements. This Certificate entitles the manufacturer to affix the CE mark to its medical devices after having prepared and signed a related EC Declaration of Conformity.
As a general rule, demonstration of conformity of medical devices and their manufacturers with the essential requirements must be based, among other things, on the evaluation of clinical data supporting the safety and performance of the products during normal conditions of use. Specifically, a manufacturer must demonstrate that the device achieves its intended performance during normal conditions of use, that the known and foreseeable risks, and any adverse events, are minimized and acceptable when weighed against the benefits of its intended performance, and that any claims made about the performance and safety of the device are supported by suitable evidence.
All manufacturers placing medical devices into the market in the EEA must comply with the EU Medical Device Vigilance System. Under this system, incidents must be reported to the relevant authorities of the member states of the EEA, and manufacturers are required to take Field Safety Corrective Actions, or FSCAs, to reduce a risk of death or serious deterioration in the state of health associated with the use of a medical device that is already placed on the market. An incident is defined as any malfunction or deterioration in the characteristics and/or performance of a device, as well as any inadequacy in the labeling or the instructions for use which, directly or indirectly, might lead to or might have led to the death of a patient or user or of other persons or to a serious deterioration in their state of health. An FSCA may include the recall, modification, exchange, destruction or retrofitting of the device. FSCAs must be communicated by the manufacturer or its legal representative to its customers and/or to the end users of the device through Field Safety Notices. Where appropriate, our products commercialized in Europe are CE marked and classified as either Class I or Class II.
On April 5, 2017, the European Parliament passed the Medical Devices Regulation, which repeals and replaces the EU Medical Devices Directive. Unlike directives, which must be implemented into the national laws of the EEA member states, the regulations would be directly applicable (i.e., without the need for adoption of EEA member State laws implementing them) in all EEA member states and are intended to eliminate current differences in the regulation of medical devices among EEA member States. The Medical Devices Regulation, among other things, is intended to establish a uniform, transparent, predictable and sustainable regulatory framework across the EEA for medical devices and in vitro diagnostic devices and ensure a high level of safety and health while supporting innovation.
The Medical Device Regulation was meant to become applicable three years after publication (in May 2020). However, on April 23, 2020, to allow EEA national authorities, notified bodies, manufacturers and other actors to focus fully on urgent priorities related to the COVID-19 pandemic, the European Council and Parliament adopted Regulation 2020/561, postponing the date of application of the Medical Device Regulation by one year (to May 2021). Once applicable, the new regulations will among other things:
strengthen the rules on placing devices on the market and reinforce surveillance once they are available;
establish explicit provisions on manufacturers' responsibilities for the follow-up of the quality, performance and safety of devices placed on the market;
improve the traceability of medical devices throughout the supply chain to the end-user or patient through a unique identification number;
set up a central database to provide patients, healthcare professionals and the public with comprehensive information on products available in the EU; and
strengthen rules for the assessment of certain high-risk devices, such as implants, which may have to undergo an additional check by experts before they are placed on the market.
These modifications may have an impact on the way we design and manufacture products and the way we conduct our business in the EEA. We are progressing in our plans to meet the new requirements.
Other regulatory bodies
Our devices are sold in multiple countries and often need to be registered with local regulatory bodies such as the Therapeutic Goods Administration in Australia, Health Canada in Canada and CFDA in China.
Other Healthcare Laws
We are subject to a number of laws and regulations that may restrict our business practices, including, without limitation, anti-kickback, false claims, physician payment transparency and data privacy and security laws. The government has interpreted these laws broadly to apply to the marketing and sales activities of manufacturers and distributors like us.
The federal Anti-Kickback Statute prohibits, among other things, persons or entities from knowingly and willfully soliciting, receiving, offering or providing remuneration, directly or indirectly, in cash or in kind, in exchange for or to induce either the referral of an individual for, or the purchase, lease, order or recommendation of, any good, facility, item or service for which payment may be made, in whole or in part, under federal healthcare programs such as Medicare and Medicaid. In addition, a person or entity does not need to have actual knowledge of this statute or specific intent to violate it in order to have committed a violation. Violations of the federal Anti-Kickback Statute may result in significant civil monetary penalties for each violation, plus up to three times the remuneration involved. Violations of the Federal Anti-Kickback Statute can also result in criminal penalties, including significant criminal fines and imprisonment. In addition, violations can result in debarment, suspension or exclusion from participation in government healthcare programs, including Medicare and Medicaid.
The federal civil False Claims Act prohibits, among other things, any person or entity from knowingly presenting, or causing to be presented, a false or fraudulent claim for payment or approval to the federal government or knowingly making, using or causing to be made or used a false record or statement material to a false or fraudulent claim to the federal government. A claim includes “any request or demand” for money or property presented to the U.S. government. The civil False Claims Act also applies to false submissions that cause the government to be paid less than the amount to which it is entitled, such as a rebate. Intent to deceive is not required to establish liability under the civil False Claims Act. In addition, a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. When an entity is determined to have violated the federal civil False Claims Act, the government may impose significant civil fines and penalties for each false claim, plus treble damages, and exclude the entity from participation in Medicare, Medicaid and other federal healthcare programs. Private suits filed under the civil False Claims Act, known as qui tam actions, can be brought by individuals on behalf of the government. These individuals may share in any amounts paid by the entity to the government in fines or settlement.
The federal Civil Monetary Penalties Law prohibits, among other things, the offering or transfer of remuneration to a Medicare or state healthcare program beneficiary if the person knows or should know it is likely to influence the beneficiary’s selection of a particular provider, practitioner, or supplier of services reimbursable by Medicare or a state healthcare program, unless an exception applies.
Additionally, there has been a recent trend of increased federal and state regulation of payments and transfers of value provided to healthcare professionals or entities.
The federal Physician Sunshine Act, which requires certain manufacturers of drugs, biologicals, and medical devices or supplies that require premarket approval by or notification to the FDA, and for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, to report annually to the CMS information related to (i) payments and other transfers of value to teaching hospitals, physicians (as defined by statute) and, beginning in 2022, physician assistants, nurse practitioners and other practitioners, and (ii) ownership and investment interests held by such providers and their immediate family members. Applicable manufacturers are required to submit annual reports to CMS. Failure to submit required information may result in significant civil monetary penalties for each failure and additional penalties for “knowing failures”, for all payments, transfers of value or ownership or investment interests that are not timely, accurately, and completely reported in an annual submission, and may result in liability under other federal laws or regulations. Certain states also mandate implementation of commercial compliance programs, impose restrictions on device manufacturer marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to healthcare professionals and entities.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created federal criminal statutes that prohibit among other actions, knowingly and willfully executing, or attempting to execute, a scheme to defraud any healthcare benefit program, including private third-party payors, knowingly and willfully embezzling or stealing from a healthcare benefit program, willfully obstructing a criminal investigation of a healthcare offense, and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Like the Anti-Kickback Statute, a person or entity does not need to have actual knowledge of these statutes or specific intent to violate them in order to have committed a violation.
Also, many U.S. states and countries outside the U.S. have similar fraud and abuse statutes or regulations that may be broader in scope and may apply regardless of payor, in addition to items and services reimbursed under government programs.
Under HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act of 2009, or HITECH, which we collectively refer to as HIPAA, the Department of Health and Human Services, or HHS, has issued regulations, including the HIPAA Privacy, Security and Breach Notification Rules, to protect the privacy and security of protected health information, or PHI, used or disclosed by covered entities including health care providers and their business associates. HIPAA also regulates standardization of data content, codes and formats used in health care transactions and standardization of identifiers for health plans and providers. Penalties for violations of HIPAA regulations include significant civil and criminal penalties for each violation. In addition to federal privacy and security regulations, there are a number of state laws governing confidentiality and security of personally identifiable information that are applicable to our business. For example, the California Consumer Privacy Act, or the CCPA, became effective on January 1, 2020. The CCPA gives California residents expanded rights to access and delete their personal information, opt out of certain personal information sharing and receive detailed information about how their personal information is used by requiring covered companies to provide new disclosures to California consumers (as that term is broadly defined) and provide such consumers new ways to opt-out of certain sales of personal information. The CCPA provides for civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. Although the law includes limited exceptions, including for “protected health information” maintained by a covered entity or business associate, it may regulate or impact our processing of personal information depending on the context. CCPA’s implementation standards and enforcement practices are likely to remain uncertain for the foreseeable future, and the CCPA may increase our compliance costs and potential liability. Similar privacy laws have been proposed at the federal level and in other states.
In some of our operations, such as those involving our cloud-based software digital health applications, we are a business associate under HIPAA and therefore required to comply with the HIPAA Security Rule, Breach Notification Rule and certain provisions of the HIPAA Privacy Rule, as well as the terms of our business associate agreements that we enter into with our covered entity customers, and are subject to significant civil and criminal penalties for failure to do so.
In addition, the General Data Protection Regulation, or GDPR, went into effect in May 2018. The GDPR imposes stringent data protection requirements for the processing of personal data in the European Economic Area, or EEA. The GDPR imposes several stringent requirements for controllers and processors of personal data, and has increased our obligations, for example, by imposing higher standards for obtaining consent from individuals to process their personal data, requiring more robust disclosures to individuals, strengthening individual data rights, shortening timelines for data breach notifications, limiting retention periods and secondary use of information (including for research purposes), increasing requirements pertaining to health data and pseudonymised (i.e., key-coded) data and imposing additional obligations when we contract with third party processors in connection with the processing of the personal data. The GDPR also imposes strict rules on the transfer of personal data out of the EEA, including to the United States; recent legal developments in Europe have created complexity and uncertainty regarding such transfers of personal data from the EEA to the United States. For example, on July 16, 2020, the Court of Justice of the European Union, or CJEU, invalidated the EU-US Privacy Shield Framework, or Privacy Shield, under which personal data could be transferred from the EEA to United States entities that had self-
certified under the Privacy Shield scheme. While the CJEU upheld the adequacy of the standard contractual clauses (a standard form of contract approved by the European Commission as an adequate personal data transfer mechanism, and potential alternative to the Privacy Shield), it made clear that reliance on them alone may not necessarily be sufficient in all circumstances. Use of the standard contractual clauses must now be assessed on a case-by-case basis taking into account the legal regime applicable in the destination country, in particular applicable surveillance laws and rights of individuals and additional measures and/or contractual provisions may need to be put in place, however, the nature of these additional measures is currently uncertain. European data protection law provides that EEA member states may make their own further laws and regulations limiting the processing of genetic, biometric or health data, which could limit our ability to use and share personal data or could cause our costs could increase, and harm our business and financial condition. The GDPR and other similar regulations impose additional conditions in order to satisfy such consent for electronic marketing, such as a prohibition on pre-checked tick boxes and bundled consents, thereby requiring customers to affirmatively consent for a given purpose through separate tick boxes or other affirmative action. Failure to comply with the requirements of GDPR and the applicable national data protection and marketing laws of the EEA member states may result in fines of up to €20,000,000 or up to 4% of the total worldwide annual turnover of the preceding financial year, whichever is higher, and other administrative penalties as well as individual claims for compensation.
Further, following the United Kingdom’s departure from the EU and EEA on January 31, 2020 and the end of the transition period on December 31, 2020, we will have to comply with the GDPR and the GDPR as incorporated into the United Kingdom domestic law, the Data Protection Act 2018, the latter regime having the ability to separately fine up to the greater of £17.5 million or 4% of global turnover. Compliance with these and any other applicable privacy and data security laws and regulations is a rigorous and time-intensive process, and we may be required to put in place additional mechanisms ensuring compliance with the new data protection rules. If we fail to comply with any such laws or regulations, we may face significant fines and penalties that could adversely affect our business, financial condition and results of operations.
Numerous other state, federal and foreign laws, including consumer protection laws and regulations, govern the collection, dissemination, use, access to, confidentiality and security of patient health information. In addition, Congress and some states are considering new laws and regulations that further protect the privacy and security of medical records or medical information. With the recent increase in publicity regarding data breaches resulting in improper dissemination of consumer information, all 50 states have passed laws regulating the actions that a business must take if it experiences a data breach, such as prompt disclosure to affected customers. Generally, these laws are limited to electronic data and make some exemptions for smaller breaches. Congress has also been considering similar federal legislation relating to data breaches. The Federal Trade Commission, or FTC, and states’ Attorneys General have also brought enforcement actions and prosecuted some data breach cases as unfair and/or deceptive acts or practices under the FTC Act. In addition to data breach notification laws, some states have enacted statutes and rules requiring businesses to reasonably protect certain types of personal information they hold or to otherwise comply with certain specified data security requirements for personal information. These laws may apply directly to our business or indirectly by contract when we provide services to other companies. We intend to continue to comprehensively protect all personal information and to comply with all applicable laws regarding the protection of such information.
The shifting commercial compliance environment and the need to build and maintain robust systems to comply with different compliance or reporting requirements in multiple jurisdictions increase the possibility that a healthcare company may fail to comply fully with one or more of these requirements. If our operations are found to be in violation of any of the health regulatory laws described above or any other laws that apply to us, we may be subject to penalties, including potentially significant criminal and civil and administrative penalties, damages, fines, disgorgement, imprisonment, exclusion from participation in government healthcare programs, contractual damages, reputational harm, administrative burdens, diminished profits and future earnings, and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our results of operations.
Employees
As of June 30, 2020, we had approximately 7,770 employees or full-time consultants, of which approximately 3,490 were employed in cost of sales activities including areas such as warehousing and manufacturing, 1,280 in research and development and 3,000 in sales, marketing and administration. Of our employees and consultants, approximately 3,030 were located in the United States, Canada and Latin America, 1,560 in Australia, 1,260 in Europe and 1,920 in Asia. We believe that the success of our business will depend, in part, on our ability to attract and retain qualified personnel.
ITEM 1A RISK FACTORS
Before deciding to purchase, hold or sell our common stock, you should carefully consider the risks described below in addition to the other cautionary statements and risks described elsewhere, and the other information contained, in this Report and in our other filings with the SEC, including our subsequent reports on Forms 10-Q and 8-K. The risks and uncertainties described below are not the only ones we face. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also affect our business. If any of these known or unknown risks or uncertainties actually occurs with material adverse effects on us, our business, financial condition and results of operations could be seriously harmed. In that event, the market price for our common stock will likely decline, and you may lose all or part of your investment.
Our inability to compete successfully in our markets may harm our business. The markets for our products, which encompass Sleep and Respiratory Care products and SaaS offerings, are highly competitive and are characterized by frequent product improvements and evolving technology. Our ability to compete successfully depends, in part, on our ability to develop, manufacture and market innovative new products. For our Sleep and Respiratory Care business, the development of innovative new products by our competitors or the discovery of alternative treatments or potential cures for the conditions that our products treat could make our products noncompetitive or obsolete. Current competitors, new entrants, academics, and others are trying to develop new devices, alternative treatments or cures, and pharmaceutical solutions to the conditions our products treat. For SaaS, the market for business management software is highly competitive, rapidly evolving, subject to changing technology, with low barriers to entry, shifting customer needs and frequent introductions of new products and services. Many prospective customers have invested substantial personnel and financial resources to implement and integrate their current business management software into their operations and, therefore, may be reluctant or unwilling to change from their current solution or provider to one of our platforms or products.
Additionally, some of our competitors have greater financial, research and development, manufacturing and marketing resources than we do. The past several years have seen a trend towards consolidation in the healthcare industry and in the markets for our products. Industry consolidation could result in greater competition if our competitors combine their resources, if our competitors are acquired by other companies with greater resources than ours, or if our competitors become affiliated with customers of ours. This competition could increase pressure on us to reduce the selling prices of our products or could cause us to increase our spending on research and development and sales and marketing. If we are unable to develop innovative new products, maintain competitive pricing, and offer products that consumers perceive to be as good as those of our competitors, our sales or gross margins could decrease which would harm our business.
Our business depends on our ability to market effectively to dealers of home healthcare products and sleep clinics. We market our products primarily to home healthcare dealers and to sleep clinics that diagnose OSA and other sleep disorders, as well as to non-sleep specialist physician practices that diagnose and treat sleep disorders. We believe that these groups play a significant role in determining which brand of product a patient will use. The success of our business depends on our ability to market effectively to these groups to ensure that our products are properly marketed and sold by these third-parties.
We have limited resources to market to the sleep clinics, home healthcare dealer branch locations and to the non-sleep specialists, most of whom use, sell or recommend several brands of products. In addition, home healthcare dealers have experienced price pressures as government and third-party reimbursement has declined for home healthcare products, and home healthcare dealers are requiring price discounts and longer periods of time to pay for products purchased from us. We cannot assure you that physicians will continue to prescribe our products, or that home healthcare dealers or patients will not substitute competing products when a prescription specifying our products has been written.
We have expanded our marketing activities in some markets to target the population with a predisposition to sleep-disordered breathing as well as primary care physicians and various medical specialists. We cannot assure you that these marketing efforts will be successful in increasing awareness or sales of our products.
Consolidation in the health care industry could have an adverse effect on our revenues and results of operations. Many home health care dealers and out-of-hospital health providers are consolidating, which may result in greater concentration of market power. As the health care industry consolidates, competition to provide goods and services to industry participants may become more intense. These industry participants may try to use their market power to negotiate price concessions or reductions for medical devices and components produced by us. If we are forced to reduce our prices because of consolidation in the health care industry, our revenues may decrease and our consolidated earnings, financial condition, and/or cash flows may suffer.
If we are unable to support our continued growth, our business could suffer. As we continue to grow, the complexity of our operations increases, placing greater demands on our management. Our ability to manage our growth effectively depends on our ability to implement and improve our financial and management information systems on a timely basis and to effect other changes in our business including, the ability to monitor and improve manufacturing systems, information technology, and quality and regulatory compliance systems, among others. Unexpected difficulties during expansion, the failure to attract and retain qualified employees, the failure to successfully replace or upgrade our management information systems, the failure to manage costs or our inability to respond effectively to growth or plan for future expansion could cause our growth to stop. If we fail to manage our growth effectively and efficiently, our costs could increase faster than our revenues and our business results could suffer.
Our business, financial condition and results of operations could be harmed by the effects of the COVID-19 pandemic. We are subject to risks related to the global pandemic associated with COVID-19, which may have an adverse impact on certain aspects of our business. Specifically, diagnostic pathways for sleep apnea treatment, including physician practices, HME suppliers and sleep clinics, have been impacted and, in some instances, been required, or in the future may be required, to temporarily close due to governments’ “shelter-in-place” orders, quarantines or similar orders or restrictions enacted to control the spread of COVID-19. In some countries, new patients are prescribed sleep apnea treatment through hospitals that are directing their resources to critical care, including COVID-19 treatment. The impact on these diagnostic and prescription pathways has resulted and may continue to result in a decrease in demand for our products designed to treat sleep apnea.
While we have experienced increased demand for our respiratory care products due to the nature of COVID-19, we cannot guarantee that demand will continue or that we will be able to identify and obtain adequate raw materials or otherwise maintain operations, supply chains and distribution systems to satisfy demand for our products in a cost-effective manner or at all. Additionally, if the increase in demand currently being experienced for our respiratory care products declines more abruptly than expected this could adversely impact our inventory levels and may result in excess inventory, which we may be unable to sell. Furthermore, due to governments’ varying restrictions on international and domestic travel, access to labor for our manufacturing facilities could be adversely impacted.
Our SaaS business may also be affected by COVID-19 and measures taken to control the spread of COVID-19. Some of our existing and potential SaaS customers are HME distributors and, therefore, have been impacted, or may be impacted, by the same temporary business closures noted above. We also have existing and potential SaaS customers that operate care facilities and are either receiving and treating patients infected with COVID-19 or are implementing significant measures to safeguard their facilities against a potential COVID-19 outbreak. Given these challenging business conditions and the uncertain economic environment, we expect businesses will be deterred from adopting new or changing SaaS platforms, which may adversely impact our ability to engage new customers for our SaaS businesses, or expand the services used by existing customers.
Additionally, the types of restrictions enacted to control the spread of COVID-19 have resulted in most of our employees working from home, and have resulted or may result in the employees of our key suppliers and customers working from home or, as noted above, not working at all. Neither we nor our suppliers have significant experience operating with the majority of our work forces working from home and this may disrupt our standard operations or significantly hamper our products from moving through our supply chain. If we are unable to move products efficiently through the supply chain we may be unable to satisfy customer demand, which could negatively impact our results of operations.
Health regulatory agencies globally may also experience disruptions in their operations as a result of the COVID-19 pandemic. Any delay or de-prioritization of our product development activities or delay in regulatory review resulting from such disruptions could materially affect our results of operations.
In addition to existing travel restrictions, countries may continue to close borders, impose prolonged quarantines, and further restrict travel, which may also disrupt our ability to move our product by air and sea. The continued spread of COVID-19 has also led to extreme disruption and volatility in the global capital markets, which increases the cost of, and adversely impacts access to, capital and increases economic uncertainty. While we expect COVID-19 to negatively impact certain aspects of our business, given the rapid and evolving nature of the virus and the uncertainty about its impact on society and the global economy, we cannot predict the extent to which it will affect our global operations, particularly if these impacts persist or worsen over an extended period of time.
We are subject to various risks relating to international activities that could affect our overall profitability. We manufacture substantially all of our products outside the United States and sell a significant portion of our products in non-U.S. markets. Sales in combined Europe, Asia and other markets accounted for approximately 38% and 39% of our net revenues in the years ended June 30, 2020 and June 30, 2019 respectively. We expect that sales within these areas will account for approximately 35-40% of our net revenues in the foreseeable future. Our sales and operations outside of the U.S. are subject to several difficulties and risks that are separate and distinct from those we face in the U.S., including:
fluctuations in currency exchange rates;
tariffs and other trade barriers;
compliance with foreign medical device manufacturing regulations;
difficulty in enforcing agreements and collecting receivables through foreign legal systems;
reduction in third-party payor reimbursement for our products;
inability to obtain import licenses;
the impact of public health epidemics/pandemics on the global economy, such as COVID-19 that has spread globally;
changes in trade policies and in U.S. and foreign tax policies;
possible changes in export or import restrictions; and
the modification or introduction of other governmental policies with potentially adverse effects.
Any of the above factors may have a material adverse effect on our ability to increase or maintain our non-U.S. sales.
If we fail to effectively integrate and capitalize on our acquisitions, combining them with our other SaaS operations, our SaaS businesses could suffer. Part of our growth strategy includes acquiring businesses consistent with our commitment to innovation in developing products for the diagnosis and treatment of sleep apnea and respiratory care as well as our SaaS business. For example, we acquired MatrixCare in November 2018 and Propeller Health in January 2019. The success of our acquisitions will depend, in part, on our ability to successfully integrate the business and operations of the acquired companies. Additionally, our management may have their attention diverted while trying to integrate these businesses. If we are not able to successfully integrate the operations, we may not realize the anticipated benefits of the acquisitions fully or at all, or may take longer to realize than expected.
We have made certain assumptions relating to our recent acquisitions that may prove to be materially inaccurate. We have made certain assumptions relating to our recent acquisitions, including MatrixCare, such as:
projections of each acquired company’s future revenue;
the amount of goodwill and intangibles that will result from our acquisitions;
acquisition costs, including transaction, contingent consideration and integration costs; and
other financial and strategic rationales and risks of the acquisitions.
While management has made such assumptions in good faith and believes them to be reasonable, the assumptions may turn out to be materially inaccurate, including for reasons beyond our control. If these assumptions are incorrect we may change or modify our assumptions, and such change or modification could have a material adverse effect on our financial condition or results of operations.
Our SaaS business depends substantially on customers entering into, renewing, upgrading and expanding their agreements for cloud services, term licenses, and maintenance and support agreements with us. Any decline in our customer renewals, upgrades or expansions could adversely affect our future operating results. We typically enter into term-based agreements for our licensed on-premises offerings, cloud services, and maintenance and support services, which customers have discretion to renew or terminate at the end of the initial term. In order for us to improve our operating results, it is important that new customers enter into renewable agreements, and our existing customers renew, upgrade and expand their term-based agreements when the initial contract term expires. Our customers have no obligation to renew, upgrade or expand their agreements with us after the terms have expired. Our customers’ renewal, upgrade and expansion rates may decline or fluctuate as a result of a number of factors, including their satisfaction or dissatisfaction with our offerings, our pricing, the effects of general economic conditions, competitive offerings or alterations or reductions in our customers’ spending levels. If our customers do not renew, upgrade or expand their agreements with us or renew on terms less favorable to us, our revenues may decline.
Government and private insurance plans may not adequately reimburse our customers for our products, which could result in reductions in sales or selling prices for our products. Our ability to sell our products depends in large part on the extent to which coverage and adequate reimbursement for our products will be available from government health administration authorities, private health insurers and other organizations. These third-party payers are increasingly challenging the prices charged for medical products and services and can, without notice, deny coverage for our products or treatments that may include the use of our products. Therefore, even if a product is approved for marketing, we cannot make assurances that coverage and reimbursement will be available for the product, that the reimbursement amount will be adequate or that the reimbursement amount, even if initially adequate, will not be subsequently reduced. For example, in some markets, such as Spain, France and Germany, government coverage and reimbursement are currently available for the purchase or rental of our products but are subject to constraints such as price controls or unit sales limitations. In other markets, such as Australia, there is currently limited or no reimbursement for devices that treat sleep apnea conditions. As we continue to develop new products, those products will generally not qualify for coverage and reimbursement until they are approved for marketing, if at all.
In the United States, we sell our products primarily to home healthcare dealers, hospitals and sleep clinics. Reductions in reimbursement to our customers by third-party payers, if they occur, may have a material impact on our customers and, therefore, may indirectly affect our pricing and sales to, or the collectability of receivables we have from, those customers. A development negatively affecting reimbursement stems from the Medicare competitive bidding program mandated by the Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (MMA). Under the program, our customers who provide HME must compete to offer products in designated competitive bidding areas, or CBAs. In addition, under the ACA, in 2016, CMS adjusted the prices in non-competitive bidding areas to match competitive bidding prices. CMS phased in the new rates beginning January 1, 2016, and were fully effective July 1, 2016. This program has significantly reduced the Medicare reimbursement to our customers compared with reimbursement in 2011, at the beginning of the program. The 21st Century Cures Act retroactively adjusted rates in non-bid areas to allow for the higher phase-in rates to be paid for items furnished between July 1, 2016 and December 31, 2016, rather than the lower fully-adjusted rates. Rules issued by CMS in 2018 resumed the higher phase-in rates in rural and non-contiguous non-competitive bidding areas for items furnished between June 1, 2018 and December 31, 2020. Pursuant to the CARES Act, these higher phase-in rates were extended through December 31, 2020, or through the end of the COVID-19 public health emergency, and were implemented in areas other than rural areas and noncontiguous areas for the same period. On March 7, 2019, CMS announced it would initiate a new round of competitive bidding, named Round 2021, with contracts expected to become effective on January 1, 2021, and extend through December 31, 2023. In addition to adopting new bidding processes, CMS expanded the product categories included in competitive bidding to include non-invasive ventilators, in addition to oxygen. However, due to the COVID-19 pandemic, CMS removed NIVs from Round 2021 of the DMEPOS Competitive Bidding Program. CPAP, and respiratory assist devices, and related supplies and accessories, which had been included in prior rounds of competitive bidding, remain included in Round 2021.
We cannot predict at this time the full impact the competitive bidding program and the developments in the competitive bidding program will have on our business and financial condition. If changes are made to this program in the future, it could affect amounts being recovered by our customers.
Healthcare reform may have a material adverse effect on our industry and our results of operations. In March 2010, the ACA was signed into law in the United States. The ACA made changes that significantly impacted the healthcare industry, including medical device manufacturers. One of the principal purposes of the ACA was to expand health insurance coverage to millions of Americans who were uninsured. The ACA required adults not covered by an employer or government-sponsored insurance plan to maintain health insurance coverage or pay a penalty, a provision commonly referred to as the individual mandate.
The ACA also contained a number of provisions designed to generate the revenues necessary to fund the coverage expansions. This included new fees or taxes on certain health-related industries, including medical device manufacturers. Beginning in 2013, entities that manufacture, produce or import medical devices were required to pay an excise tax in an amount equal to 2.3% of the price for which such devices are sold in the United States. This excise tax was applicable to our products that are primarily used in hospitals and sleep labs, which includes the ApneaLink, VPAP Tx, certain Respiratory Care and dental sleep products. Through a series of legislative amendments, the tax was suspended beginning in 2016, and permanently repealed effective January 1, 2020. In addition to the competitive bidding changes discussed above, the ACA also included, among other things, demonstrations to develop organizations that are paid under a new payment methodology for voluntary coordination of care by groups of providers, such as physicians and hospitals, and the establishment of a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in and conduct comparative clinical effectiveness research. The increased funding and focus on comparative clinical effectiveness research, which compares and evaluates the risks and benefits, clinical outcomes, effectiveness and appropriateness of products, may result in lower reimbursements by payors for our products and decreased profits to us.
Other federal legislative changes have been proposed and adopted since the ACA was enacted. These changes included an aggregate reduction in Medicare payments to providers of 2% per fiscal year, which went into effect on April 1, 2013. The CARES Act, which was signed into law in March 2020, suspended the payment reductions from May 1, 2020 through December 31, 2020, and extended the sequester by one additional year, through 2030. In addition, on January 2, 2013, the American Taxpayer Relief Act of 2012, was signed into law, which, among other things, further reduced Medicare payments to several providers, including hospitals, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
The full impact on our business of the ACA and other new laws is uncertain. Nor is it clear whether other legislative changes will be adopted, if any, or how such changes would affect the demand for our products. Future actions by the administration and the U.S. Congress including, but not limited to, repeal or replacement of the ACA could have a material adverse impact on our results of operations or financial condition. Additionally, all or a portion of the ACA and related subsequent legislation may be modified, repealed or otherwise invalidated through other judicial challenge. For example, on December 14, 2018, a U.S. District Court Judge in the Northern District of Texas, ruled that the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as part of the U.S. Tax Act, the remaining provisions of the ACA are invalid as well. On December 18, 2019, the U.S. Court of Appeals for the 5th Circuit upheld the District Court ruling that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. On March 2, 2020, the United States Supreme Court granted the petitions for writs of certiorari to review this case, although it remains unclear when or how the Supreme Court will rule. It is also unclear how other efforts to challenge, repeal or replace the ACA will impact the ACA or our business.
Various healthcare reform proposals have also emerged at the state level within the United States. The ACA as well as other federal and/or state healthcare reform measures that may be adopted in the future, singularly or in the aggregate, could have a material adverse effect on our business, financial condition and results of operations.
Failure to comply with anti-kickback and fraud regulations could result in substantial penalties and changes in our business operations. Although in the United States we do not provide healthcare services, submit claims for third-party reimbursement, or receive payments directly from Medicare, Medicaid or other third-party payors for our products, we are subject to healthcare fraud and abuse regulation and enforcement by federal, state and foreign governments, which could significantly impact our business. We also are subject to foreign fraud and abuse laws, which vary by country.
In the United States, the laws that may affect our ability to operate include, but are not limited to:
the federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering, or paying remuneration, directly or indirectly, in cash or in kind, in exchange for or to induce either the referral of an individual for, or the purchase, lease, order or recommendation of, any good, facility, item or service for which payment may be made, in whole or in part, under federal healthcare programs such as Medicare and Medicaid. A person or entity does not need to have actual knowledge of this statute or specific intent to violate the Anti-Kickback Statute itself to have committed a violation. The U.S. government has interpreted this law broadly to apply to the marketing and sales activities of manufacturers and distributors like us. Violations of the federal Anti-Kickback Statute may result in significant civil monetary penalties for each violation, plus up to three times the remuneration involved. Violations of the Federal Anti-Kickback Statute can also result in significant criminal penalties and imprisonment;
federal civil and criminal false claims laws and civil monetary penalty laws, that prohibit, among other things, knowingly presenting, or causing to be presented, claims for payment or approval to the federal government that are false or fraudulent, knowingly making a false statement material to an obligation to pay or transmit money or property to the federal government or knowingly concealing or knowingly and improperly avoiding or decreasing an obligation to pay or transmit money or property to the federal government. These laws may apply to manufacturers and distributors who provide information on coverage, coding, and reimbursement of their products to persons who do bill third-party payors. In addition, the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act. Violations can result in debarment, suspension or exclusion from participation in government healthcare programs, including Medicare and Medicaid. When an entity is determined to have violated the federal civil False Claims Act, the government may impose significant civil fines and penalties for each false claim, plus treble damages, and exclude the entity from participation in Medicare, Medicaid and other federal healthcare programs.
HIPAA, which created federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. A person or entity does not need to have actual knowledge of these statutes or specific intent to violate them to have committed a violation. Further, failure to comply with the HIPAA privacy and security standards can result in significant civil monetary penalties per violation and, in certain circumstances, significant criminal penalties and/or imprisonment;
the federal Physician Sunshine Act requirements under the ACA, which impose reporting and disclosure requirements on device and drug manufacturers for any “transfer of value” made or distributed by certain manufacturers of drugs, devices, biologics, and medical supplies to physicians (including doctors, dentists, optometrists, podiatrists and chiropractors), teaching hospitals, and ownership and investment interests held by physicians and their immediate family members. Beginning in 2022, applicable manufacturers also will be required to report such information regarding payments and transfers of value provided, as well as ownership and investment interests held, during the previous year to physician assistants, nurse practitioners, clinical nurse specialists, certified nurse anesthetists and certified nurse midwives;
federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm customers; and
state and foreign law equivalents of each of the above federal laws, such as state anti-kickback and false claims laws that may apply to items or services reimbursed by any third-party payor, including commercial insurers; state laws that require device companies to comply with the industry’s voluntary compliance guidelines and the relevant compliance guidance promulgated by the federal government, or otherwise restrict payments that may be made to healthcare providers and other potential referral sources; state laws that require device manufacturers to report information related to payments and other transfers of value to physicians and other healthcare providers or marketing expenditures.
The scope and enforcement of these laws are uncertain and subject to rapid change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. Federal and state enforcement bodies have recently increased their scrutiny of interactions between healthcare companies and healthcare providers, which has led to a number of investigations, prosecutions, convictions and settlements in the healthcare industry. Responding to investigations can be time-and resource-consuming and can divert management’s attention from the business. Additionally, as a result of these types of investigations, healthcare providers and entities may face litigation or have to agree to settlements that can include monetary penalties and onerous compliance and reporting requirements as part of a consent decree or corporate integrity agreement. Any such investigation or settlement could increase our costs or otherwise have an adverse effect on our business.
If our operations are found to be in violation of any of the laws described above or any other governmental regulations that apply to us now or in the future, we may be subject to penalties, including civil and criminal penalties, damages, fines, disgorgement, exclusion from governmental health care programs, additional compliance and reporting obligations, imprisonment and the curtailment or restructuring of our operations, any of which could adversely affect our ability to operate our business and our financial results.
In December 2019, we entered into a settlement agreement with the U.S. Department of Justice and the U.S. Attorneys’ Offices for the District Court of South Carolina, the Southern District of California, the Northern District of Iowa and the Eastern District of New York. The agreement resolves five lawsuits originally brought by whistleblowers under the qui tam provisions of the False Claims Act and allegations that we: (a) provided DME companies with free telephone call center services and other free patient outreach services that enabled these companies to order resupplies for their patients with sleep apnea, (b) provided sleep labs with free and below-cost positive airway pressure masks and diagnostic machines, as well as free installation of these machines, (c) arranged for, and fully guaranteed the payments due on, interest-free loans that DME supplies acquired from third-party financial institutions for the purchase of our equipment, and (d) provided non-sleep specialist physicians free home sleep testing devices referred to as “ApneaLink.” We agreed with the government to civilly resolve these matters for a payment of $39.5 million ($37.5 million to the federal government and $2 million to the various states) and we incurred additional fees and administrative costs that typically accompany such a resolution amounting to $1.1 million. The total final costs relating to these matters was $40.6 million.
Contemporaneous with the civil settlement, we also entered into a Corporate Integrity Agreement, or CIA, with the Department of Health and Human Services Office of Inspector General. The CIA requires, among other things, that we implement additional controls around our product pricing and sales and that we conduct internal and external monitoring of our arrangements with referrals sources. The settlement agreement with the government and the CIA could result in reputational harm, the curtailment or restructuring of our operations and an increase in our compliance costs, any of which could materially adversely affect our financial results and our ability to operate our business.
Our use and disclosure of individually identifiable information, including health information, is subject to federal, state and foreign privacy and security regulations, and our failure to comply with those regulations or to adequately secure the information we hold could result in significant liability or reputational harm. The privacy and security of personally identifiable information stored, maintained, received or transmitted electronically is a major issue in the United States and abroad. While we strive to comply with all applicable privacy and security laws and regulations, as well as our own posted privacy policies, legal standards for privacy, including but not limited to “unfairness” and “deception,” as enforced by the FTC and state attorneys general, continue to evolve and any failure or perceived failure to comply may result in proceedings or actions against us by government entities or others, or could cause us to lose audience and customers, which could have a material adverse effect on our business. Recently, there has been an increase in public awareness of privacy issues in the wake of revelations about the activities of various government agencies and in the number of private privacy-related lawsuits filed against companies. Concerns about our practices with regard to the collection, use, disclosure, or security of personally identifiable information or other privacy-related matters, even if unfounded and even if we are in compliance with applicable laws, could damage our reputation and harm our business.
Numerous foreign, federal and state laws and regulations govern collection, dissemination, use and confidentiality of personally identifiable health information, including (i) state privacy and confidentiality laws (including state laws requiring disclosure of breaches); (ii) HIPAA; and (iii) European and other foreign data protection laws, including the GDPR.
HIPAA establishes a set of national privacy and security standards for the protection of individually identifiable health information, or protected health information, by health plans, healthcare clearinghouses and healthcare providers that submit certain covered transactions electronically, or covered entities, and their “business associates,” which are persons or entities that perform certain services for, or on behalf of, a covered entity that involve creating, receiving, maintaining or transmitting protected health information. Certain portions of our business, such as the cloud-based software digital health applications, are subject to HIPAA as a business associate of our covered entity clients. To provide our covered entity clients with services that involve access to PHI, HIPAA requires us to enter into business associate agreements that require us to safeguard PHI in accordance with HIPAA. As a business associate, we are also directly liable for compliance with HIPAA. Penalties for violations of HIPAA regulations include civil and criminal penalties.
HIPAA authorizes state attorneys’ general to file suit under HIPAA on behalf of state residents. Courts can award damages, costs and attorneys’ fees related to violations of HIPAA in such cases. While HIPAA does not create a private right of action allowing individuals to sue us in civil court for HIPAA violations, its standards have been used as the basis for a duty of care claim in state civil suits such as those for negligence or recklessness in the misuse or breach of PHI.
HIPAA further requires business associates like us to notify our covered entity clients “without unreasonable delay and in no case later than 60 calendar days after discovery of the breach.” Covered entities must notify affected individuals “without unreasonable delay and in no case later than 60 calendar days after discovery of the breach” if their unsecured PHI is subject to an unauthorized access, use or disclosure. If a breach affects 500 patients or more, covered entities must report it to HHS and local media without unreasonable delay, and HHS will post the name of the breaching entity on its public website. If a breach affects fewer than 500 individuals, the covered entity must log it and notify HHS at least annually.
If we are unable to properly protect the privacy and security of health information entrusted to us, our solutions may be perceived as not secure, we may incur significant liabilities and customers may curtail their use of or stop using our solutions. In addition, if we fail to comply with the terms of our business associate agreements with our clients, we are liable not only contractually but also directly under HIPAA.
In addition, the California Consumer Privacy Act of 2018 or CCPA became effective on January 1, 2020. The CCPA gives California residents expanded rights to access and delete their personal information, opt out of certain personal information sharing and receive detailed information about how their personal information is used by requiring covered companies to provide new disclosures to California consumers (as that term is broadly defined) and provide such consumers new ways to opt-out of certain sales of personal information. The CCPA includes civil penalties for violations, as well as a private right of action for data breaches that is expected to increase data breach litigation. Although the law includes limited exceptions, including for “protected health information” maintained by a covered entity or business associate, it may regulate or impact our processing of personal information depending on the context. CCPA’s implementation standards and enforcement practices are likely to remain uncertain for the foreseeable future, and the CCPA may increase our compliance costs and potential liability. Any failure or perceived failure by us to comply with privacy or security laws, policies, legal obligations or industry standards or any security incident that results in the unauthorized release or transfer of personally identifiable information may also result in governmental enforcement actions and investigations, fines and penalties, litigation and/or adverse publicity, including by consumer advocacy groups, and could cause our customers to lose trust in us, which could have an adverse effect on our reputation and business. Such failures could have a material adverse effect on our financial condition and operations. If the third parties we work with violate applicable laws, contractual obligations or suffer a security breach, such violations may also put us in breach of our obligations under privacy laws and regulations and/or could in turn have a material adverse effect on our business.
We are also subject to laws and regulations in non-U.S. countries covering data privacy and the protection of health-related and other personal information. For example, EU member states and other jurisdictions have adopted data protection laws and regulations, which impose significant compliance obligations. Laws and regulations in these jurisdictions apply broadly to the collection, use, storage, disclosure and security of personal information that identifies or may be used to identify an individual, such as names, contact information, and sensitive personal data such as health data. These laws and regulations are subject to frequent revisions and differing interpretations, and have generally become more stringent over time.
In addition, the GDPR went into effect in May 2018. The GDPR imposes stringent data protection requirements for the processing of personal data in the European Economic Area, or EEA. The GDPR imposes several stringent requirements for controllers and processors of personal data, and increased our obligations, for example, by imposing higher standards for obtaining consent from individuals to process their personal data, requiring more robust disclosures to individuals, strengthening individual data rights, shortening timelines for data breach notifications, limiting retention periods and secondary use of information (including for research purposes), increasing requirements pertaining to health data and pseudonymised (i.e., key-coded) data and imposing additional obligations when we contract with third party processors in connection with the processing of the personal data. The GDPR also imposes strict rules on the transfer of personal data out of the EEA, including to the United States, and recent legal developments in Europe have created complexity and uncertainty regarding such transfers of personal data from the EEA to the United States. For example, on July 16, 2020, the Court of Justice of the European Union, or CJEU, invalidated the EU-US Privacy Shield Framework, or Privacy Shield, under which personal data could be transferred from the EEA to United States entities that had self-certified under the Privacy Shield scheme. While the CJEU upheld the adequacy of the standard contractual clauses (a standard form of contract approved by the European Commission as an adequate personal data transfer mechanism, and potential alternative to the Privacy Shield), it made clear that reliance on them alone may not necessarily be sufficient in all circumstances. Use of the standard contractual clauses must now be assessed on a case-by-case basis taking into account the legal regime applicable in the destination country, in particular applicable surveillance laws and rights of individuals and additional measures and/or contractual provisions may need to be put in place, however, the nature of these additional measures is currently uncertain. European data protection law provides that EEA member states may make their own further laws and regulations limiting the processing of genetic, biometric or health data, which could limit our ability to use and share personal data or could cause our costs could increase, and harm our business and financial condition. The GDPR and other similar regulations impose additional conditions in order to satisfy such consent for electronic marketing, such as a prohibition on pre-checked tick boxes and bundled consents, thereby requiring customers to affirmatively consent for a given purpose through separate tick boxes or other affirmative action. Failure to comply with the requirements of GDPR and the applicable national data protection and marketing laws of the EEA member states may result in fines of up to €20,000,000 or up to 4% of the total worldwide annual turnover of the preceding financial year, whichever is higher, and other administrative penalties as well as individual claims for compensation.
In addition, following the United Kingdom’s departure from the EU and the EEA on January 31, 2020 and the end of the transition period on December 31, 2020, we will have to comply with the GDPR and the GDPR as incorporated into the United Kingdom domestic law, the Data Protection Act 2018, the latter regime having the ability to separately fine up to the greater of £17.5 million or 4% of global turnover. Compliance with these and any other applicable privacy and data security laws and regulations is a rigorous and time-intensive process, and we may be required to put in place additional mechanisms ensuring compliance with the new data protection rules. If we fail to comply with any such laws or regulations, we may face significant fines and penalties that could adversely affect our business, financial condition and results of operations.
Our business activities are subject to extensive regulation, and any failure to comply could have a material adverse effect on our business, financial condition, or results of operations. We are subject to extensive U.S. federal, state, local and international regulations regarding our business activities. Failure to comply with these regulations could result in, among other things, recalls of our products, substantial fines and criminal charges against us or against our employees. Furthermore, certain of our products could be subject to recall if the Food and Drug Administration, or the FDA, other regulators or we determine, for any reason, that those products are not safe or effective. Any recall or other regulatory action could increase our costs, damage our reputation, affect our ability to supply customers with the quantity of products they require and materially affect our operating results.
Actual or attempted breaches of security, unauthorized disclosure of information, denial of service attacks or the perception that personal and/or other sensitive or confidential information in our possession is not secure, could result in a material loss of business, substantial legal liability or significant harm to our reputation. We receive, collect, process, use and store a large amount of information from clients and our own employees, including personally identifiable, protected health and other sensitive and confidential information. This data is often accessed by us through transmissions over public and private networks, including the Internet. The secure transmission of such information over the Internet and other mechanisms is essential to maintain confidence in our information technology systems. We have implemented security measures, technical controls and contractual precautions designed to identify, detect and prevent unauthorized access, alteration, use or disclosure of our clients’, patients’ and employees’ data. However, the techniques used in these attacks change frequently and may be difficult to detect for periods of time and we may face difficulties in anticipating and implementing adequate preventative measures. As a result of the COVID-19 pandemic, we may face increased cybersecurity risks due to our reliance on internet technology and the number of our employees who are working remotely, which may create additional opportunities for cybercriminals to exploit vulnerabilities. Beyond external criminal activity, systems that access or control access to our services and databases may be compromised as a result of human error, fraud or malice on the part of employees or third parties, or may result from accidental technological failure. Because the techniques used to circumvent security systems can be highly sophisticated and change frequently, often are not recognized until launched against a target and may originate from less regulated and remote areas around the world, we may be unable to proactively address all possible techniques or implement adequate preventive measures for all situations.
If someone is able to circumvent or breach our security systems, they could steal any information located therein or cause serious and potentially long lasting disruption to our operations. Security breaches or attempts thereof could also damage our reputation and expose us to a risk of monetary loss and/or litigation, fines and sanctions. We also face risks associated with security breaches affecting third parties that conduct business with us or our clients and others who interact with our data. While we maintain insurance that covers certain security and privacy breaches, we may not carry appropriate insurance or maintain sufficient coverage to compensate for all potential liability.
We are subject to diverse laws and regulations relating to data privacy and security, including HIPAA and European data privacy laws. Complying with these numerous and complex regulations is expensive and difficult, and failure to comply with these regulations could result in regulatory scrutiny, fines and civil liability. In addition, any security breach or attempt thereof could result in liability for stolen assets or information, additional costs associated with repairing any system damage, incentives offered to clients or other business partners to maintain business relationships after a breach, and implementation of measures to prevent future breaches, including organizational changes, deployment of additional personnel and protection technologies, employee training and engagement of third-party experts and consultants. Additionally, the costs incurred to remediate any data security or privacy incident could be substantial.
We cannot assure you that any of our third-party service providers with access to our or our clients and/or employees’ personally identifiable and other sensitive or confidential information will maintain appropriate policies and practices regarding data privacy and security in compliance with all applicable laws or that they will not experience data security breaches or attempts thereof, which could have a corresponding effect on our business.
If there are interruptions or performance problems associated with our technology or infrastructure, our existing SaaS customers may experience service outages, and our new customers may experience delays in the deployment of our platform. We depend on services from various third parties as well as our own technical operations infrastructure to distribute our SaaS products via the Internet. If a service provider fails to provide sufficient capacity to support our platform or otherwise experiences service outages, such failure could interrupt our customers’ access to our service, which could adversely affect their perception of our platform's reliability and our revenues. Any disruptions in these services, including as a result of actions outside of our control, would significantly impact the continued performance of our SaaS products. In the future, these services may not be available to us on commercially reasonable terms, or at all. Any loss of the right to use any of these services could result in decreased functionality of our SaaS products until equivalent technology is either developed by us or, if available from another provider, is identified, obtained and integrated into our infrastructure.
To meet our business needs, we must maintain sufficient excess capacity in our operations infrastructure to ensure that our SaaS products are accessible. Design and mechanical errors, spikes in usage volume and failure to follow system protocols and procedures could cause our systems to fail, resulting in interruptions in our SaaS products. Any interruptions or delays in our service, whether or not caused by our products, or as a result of third-party error, our own error, natural disasters or security breaches, whether accidental or willful, could harm our relationships with customers and cause our revenue to decrease and/or our expenses to increase.
Any of the above circumstances or events may harm our reputation, cause customers to terminate their agreements with us, impair our ability to obtain contract renewals from existing customers, impair our ability to grow our customer base, result in the expenditure of significant financial, technical and engineering resources, subject us to financial penalties and liabilities under our service level agreements, and otherwise harm our business, results of operations and financial condition.
Product sales, introductions or modifications may be delayed or canceled as a result of FDA regulations or similar foreign regulations, which could cause our sales and profits to decline. Unless a product is exempt, before we can market or sell a new medical device in the United States, we must obtain FDA clearance or approval, which can be a lengthy and time-consuming process. We generally receive clearance from the FDA to market our products in the United States under Section 510(k) of the Federal Food, Drug, and Cosmetic Act or our products are exempt from the Section 510(k) clearance process. The 510(k) clearance process can be expensive, time-consuming and uncertain. In the 510(k) clearance process, the FDA must determine that a proposed device is “substantially equivalent” to a device legally on the market, known as a “predicate” device, with respect to intended use, technology and safety and effectiveness, in order to clear the proposed device for marketing. The FDA has a high degree of latitude when evaluating submissions and may determine that a proposed device submitted for 510(k) clearance is not substantially equivalent to a predicate device. After a device receives 510(k) premarket notification clearance from the FDA, any modification that could significantly affect its safety or effectiveness, or that would constitute a major change in the intended use of the device, technology, materials, packaging, and certain manufacturing processes may require a new 510(k) clearance or premarket approval. We have modified some of our Section 510(k) approved products without submitting new Section 510(k) notices, which we do not believe were required. However, if the FDA disagrees with us and requires us to submit new Section 510(k) notifications for modifications to our existing products, we may be required to stop marketing the products while the FDA reviews the Section 510(k) notification.
Any new product introduction or existing product modification could be subjected to a lengthier, more rigorous FDA examination process. For example, in certain cases we may need to conduct clinical trials of a new product before submitting a 510(k) notice. We may also be required to obtain premarket approvals for certain of our products. Indeed, recent trends in the FDA’s review of premarket notification submissions suggest that the FDA is often requiring manufacturers to provide new, more expansive, or different information regarding a particular device than what the manufacturer anticipated upon 510(k) submission. This has resulted in increasing uncertainty and delay in the premarket notification review process. For example, in November 2018, FDA officials announced forthcoming steps that the FDA intends to take to modernize the 510(k) premarket notification pathway. Among other things, the FDA announced that it plans to develop proposals to drive manufacturers utilizing the 510(k) pathway toward the use of newer predicates. These proposals include plans to potentially sunset certain older devices that were used as predicates under the 510(k) clearance pathway, and to potentially publish a list of devices that have been cleared on the basis of demonstrated substantial equivalence to predicate devices that are more than 10 years old. In September 2019, the FDA also issued revised final guidance establishing a “Safety and Performance Based Pathway” for “manufacturers of certain well-understood device types” allowing manufacturers to rely on objective safety and performance criteria recognized by the FDA to demonstrate substantial equivalence, obviating the need for manufacturers to compare the safety and performance of their medical devices to specific predicate devices in the clearance process. The FDA intends to develop and maintain a list of device types appropriate for the “safety and performance based” pathway and will continue to develop product-specific guidance documents that identify the performance criteria and recommended testing methodologies for each such device type, where feasible. Some of these proposals have not yet been finalized or adopted, and the FDA announced that it would seek public feedback prior to publication of any such proposals, and may work with Congress to implement such proposals through legislation. Accordingly, it is unclear the extent to which any proposals, if adopted, could impose additional regulatory requirements on us that could delay our ability to obtain new 510(k) clearances, increase the costs of compliance, or restrict our ability to maintain our current clearances, or otherwise create competition that may negatively affect our business.
The FDA’s ongoing review of the 510(k) program may make it more difficult for us to make modifications to our previously cleared products, either by imposing stricter requirements on when a manufacturer must submit a new 510(k) for a modification to a previously cleared product, or by applying more onerous review criteria to such submissions. FDA continues to review its 510(k) clearance process which could result in additional changes to regulatory requirements or guidance documents which could increase the costs of compliance, or restrict our ability to maintain current clearances. The requirements of the more rigorous premarket approval process and/or significant changes to the 510(k) clearance process could delay product introductions and increase the costs associated with FDA compliance. Marketing and sale of our products outside the United States are also subject to regulatory clearances and approvals, and if we fail to obtain these regulatory approvals, our sales could suffer. We cannot assure you that any new products we develop will receive required regulatory approvals from U.S. or foreign regulatory agencies.
We are subject to substantial regulation related to quality standards applicable to our manufacturing and quality processes. Our failure to comply with these standards could have an adverse effect on our business, financial condition, or results of operations. The FDA regulates the approval, manufacturing, and sales and marketing of many of our products in the United States. Significant government regulation also exists in Canada, Japan, Europe, and other countries in which we conduct business. As a device manufacturer, we are required to register with the FDA and are subject to periodic inspection by the FDA for compliance with the FDA’s Quality System Regulation requirements, which require manufacturers of medical devices to adhere to certain regulations, including testing, quality control and documentation procedures. In addition, the federal Medical Device Reporting regulations require us to provide information to the FDA whenever there is evidence that reasonably suggests that a device may have caused or contributed to a death or serious injury or, if a malfunction were to occur, could cause or contribute to a death or serious injury. Compliance with applicable regulatory requirements is subject to continual review and is rigorously monitored through periodic inspections by the FDA. In the European Community, we are required to maintain certain ISO certifications in order to sell our products and must undergo periodic inspections by notified bodies to obtain and maintain these certifications. Failure to comply with current governmental regulations and quality assurance guidelines could lead to temporary manufacturing shutdowns, product recalls or related field actions, product shortages or delays in product manufacturing. Efficacy or safety concerns, an increase in trends of adverse events in the marketplace, and/or manufacturing quality issues with respect to our products could lead to product recalls or related field actions, withdrawals, and/or declining sales.
Disruptions at the FDA and other government agencies caused by funding shortages or global health concerns could hinder their ability to hire, retain or deploy key leadership and other personnel, or otherwise prevent new or modified products from being developed, cleared or approved or commercialized in a timely manner or at all, which could negatively impact our business. The ability of the FDA to review and clear or approve new products can be affected by a variety of factors, including government budget and funding levels, statutory, regulatory, and policy changes, the FDA’s ability to hire and retain key personnel and accept the payment of user fees, and other events that may otherwise affect the FDA’s ability to perform routine functions. Average review times at the FDA have fluctuated in recent years as a result. In addition, government funding of other government agencies that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable. Disruptions at the FDA and other agencies may also slow the time necessary for medical devices or modifications to cleared or approved medical devices to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, including for 35 days beginning on December 22, 2018, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA, have had to furlough critical FDA employees and stop critical activities.
Separately, in response to the COVID-19 pandemic, on March 10, 2020, the FDA announced its intention to postpone most foreign inspections of manufacturing facilities, and subsequently, on March 18, 2020, the FDA temporarily postponed routine surveillance inspections of domestic manufacturing facilities. Regulatory authorities outside the United States may adopt similar restrictions or other policy measures in response to the COVID-19 pandemic. Subsequently, on July 10, 2020, the FDA announced its intention to resume certain on-site inspections of domestic manufacturing facilities subject to a risk-based prioritization system. The FDA intends to use this risk-based assessment system to identify the categories of regulatory activity that can occur within a given geographic area, ranging from mission critical inspections to resumption of all regulatory activities. If a prolonged government shutdown occurs, or if global health concerns continue to prevent the FDA or other regulatory authorities from conducting their regular inspections, reviews, or other regulatory activities, it could significantly impact the ability of the FDA or other regulatory authorities to timely review and process our regulatory submissions, which could have a material adverse effect on our business.
Laws regulating consumer contacts could adversely affect our business operations or create liabilities. Our business activities include contacts with consumers in different parts of the world. Certain laws, such as the U.S. Telephone Consumer Protection Act, regulate telemarketing practices and certain automated outbound contacts with consumers, such as phone calls, texts or emails. Our use of outbound contacts may be restricted by existing laws, or by laws, regulations, or regulatory decisions that may be adopted in the future. Similarly, the new California Consumer Privacy Act of 2018 requires disclosure of our privacy practices to consumers. If we are found to have violated these laws or regulations, we may be subjected to substantial fines, penalties, or liabilities to consumers.
Our products are the subject of clinical trials conducted by us, our competitors, or other third parties, the results of which may be unfavorable, or perceived as unfavorable, and could have a material adverse effect on our business, financial condition, and results of operations. As a part of the regulatory process to obtain marketing clearance for new products and new indications for existing products, or for other reasons, we conduct and participate in numerous clinical trials with a variety of study designs, patient populations, and trial endpoints. We, our competitors, or other third parties may also conduct clinical trials involving our commercially marketed products. The results of clinical trials may be unfavorable or inconsistent with previous findings, or could identify safety signals associated with our products. Current or future clinical trials may not meet primary endpoints, may reveal disadvantages of our products and solutions for various markets we address, or could generate unfavorable or inconsistent clinical data. Clinical data, or the market’s or regulatory bodies’ perception of the clinical data, may adversely impact our ability to obtain product clearances or approvals, and our position in, and share of, the markets in which we participate. Moreover, if these clinical trials identify serious safety issues associated with our marketed products, potentially adverse consequences could result, including that regulatory authorities could withdraw clearances or approvals of our products, we could be required to halt the marketing and sales of our products or recall our products, we could be required to update our product labeling with additional warnings, we could be sued and held liable for harm caused to patients, and our reputation may suffer. Any of these could have a material adverse impact on our business, financial condition, and results of operations.
Off-label marketing of our products could result in substantial penalties. The FDA strictly regulates the promotional claims that may be made about FDA-cleared products. In particular, clearance under Section 510(k) only permits us to market our products for the uses indicated on the labeling cleared by the FDA. We may request additional label indications for our current products, and the FDA may deny those requests outright, require additional expensive clinical data to support any additional indications or impose limitations on the intended use of any cleared products as a condition of clearance. If the FDA determines that we have marketed our products for off-label use, we could be subject to fines, injunctions or other penalties. It is also possible that other federal, state or foreign enforcement authorities might take action if they consider our business activities to constitute promotion of an off-label use, which could result in significant penalties, including, but not limited to, criminal, civil and administrative penalties, damages, fines, disgorgement, exclusion from participation in government healthcare programs, and the curtailment of our operations. Any of these events could significantly harm our business and results of operations and cause our stock price to decline.
Disruptions in the supply of components from our suppliers could result in a significant reduction in sales and profitability. We purchase configured components for our devices from various suppliers, including some who are single-source suppliers for us. Disruptions to our suppliers, including disruptions in connection with the novel strain of coronavirus (COVID-19), may limit our ability to manufacture our devices in a timely or cost-effective manner, which could result in a significant reduction in sales and profitability. We cannot assure you that a replacement supplier would be able to configure its components for our devices on a timely basis or, in the alternative, that we would be able to reconfigure our devices to integrate the replacement part. A reduction or halt in supply while a replacement supplier reconfigures its components, or while we reconfigure our devices for the replacement part, would limit our ability to manufacture our devices in a timely or cost-effective manner, which could result in a significant reduction in sales and profitability. We cannot assure you that our inventories would be adequate to meet our production needs during any prolonged interruption of supply.
If we fail to attract develop and retain key employees our business may suffer. Our ability to compete effectively depends on our ability to attract and retain key employees, including people in senior management, sales, marketing, technology and R&D positions. Competition for top talent in the healthcare industry can be intense. Our ability to recruit and retain such talent will depend on a number of factors, including hiring practices of our competitors, compensation and benefits, work location, work environment and industry economic conditions. If we cannot effectively recruit, develop and retain qualified employees to drive our strategic goals, our business could suffer.
We are subject to potential product liability claims that may exceed the scope and amount of our insurance coverage, which would expose us to liability for uninsured claims. We are subject to potential product liability claims as a result of the design, manufacture and marketing of medical devices. Any product liability claim brought against us, with or without merit, could result in the increase of our product liability insurance rates. In addition, we would have to pay any amount awarded by a court in excess of our policy limits. Our insurance policies have various exclusions, and thus we may be subject to a product liability claim for which we have no insurance coverage, in which case, we may have to pay the entire amount of any award. We cannot assure you that our insurance coverage will be adequate or that all claims brought against us will be covered by our insurance and we cannot assure you that we will be able to obtain insurance in the future on terms acceptable to us or at all. A successful product liability claim brought against us in excess of our insurance coverage, if any, may require us to pay substantial amounts, which could harm our business.
If our SaaS products fail to perform properly and if we fail to develop enhancements, we could lose customers, become subject to service performance or warranty claims and our market share could decline. Our SaaS operations are dependent upon our ability to prevent system interruptions and, as we continue to grow, we will need to devote additional resources to improving our infrastructure in order to maintain the performance of our products and solutions. The applications underlying our SaaS products are inherently complex and may contain material defects or errors, which may cause disruptions in availability or other performance problems. We have from time to time found defects in our products and may discover additional defects in the future that could result in data unavailability, unauthorized access to, loss, corruption or other harm to our customers’ data. While we implement bug fixes and upgrades as part of our regularly scheduled system maintenance, we may not be able to detect and correct defects or errors before implementing our products and solutions. Consequently, we or our customers may discover defects or errors after our products and solutions have been deployed. If we fail to perform timely maintenance or if customers are otherwise dissatisfied with the frequency and/or duration of our maintenance services and related system outages, our existing customers could elect not to renew their contracts, delay or withhold payment, or potential customers may not adopt our products and solutions and our brand and reputation could be harmed. In addition, the occurrence of any material defects, errors, disruptions in service or other performance problems with our software could result in warranty or other legal claims against us and diversion of our resources. The costs incurred in addressing and correcting any material defects or errors in our software and expanding our infrastructure and architecture in order to accommodate increased demand for our products and solutions may be substantial and could adversely affect our operating results.
Our intellectual property may not protect our products, and/or our products may infringe on the intellectual property rights of third-parties. We rely on a combination of patents, trade secrets and non-disclosure agreements to protect our intellectual property. Our success depends, in part, on our ability to obtain and maintain United States and foreign patent protection for our products, their uses and our processes to preserve our trade secrets and to operate without infringing on the proprietary rights of third-parties. We have a number of pending patent applications, and we do not know whether any patents will issue from any of these applications. We do not know whether any of the claims in our issued patents or pending applications will provide us with any significant protection against competitive products or otherwise be commercially valuable. Legal standards regarding the validity of patents and the proper scope of their claims are still evolving, and there is no consistent law or policy regarding the valid breadth of claims. Additionally, there may be third-party patents, patent applications and other intellectual property relevant to our products and technology which are not known to us and that block or compete with our products. We face the risks that:
third-parties will infringe our intellectual property rights;
our non-disclosure agreements will be breached;
we will not have adequate remedies for infringement;
our trade secrets will become known to or independently developed by our competitors; or
third-parties will be issued patents that may prevent the sale of our products or require us to license and pay fees or royalties in order for us to be able to market some of our products.
Litigation may be necessary to enforce patents issued to us, to protect our proprietary rights, or to defend third-party claims that we have infringed on proprietary rights of others. If the outcome of any litigation or proceeding brought against us were adverse, we could be subject to significant liabilities to third-parties, could be required to obtain licenses from third-parties, could be forced to design around the patents at issue or could be required to cease sales of the affected products. A license may not be available at all or on commercially viable terms, and we may not be able to redesign our products to avoid infringement. Additionally, the laws regarding the enforceability of patents vary from country to country, and we cannot assure you that any patent issues we face will be uniformly resolved, or that local laws will provide us with consistent rights and benefits.
Tax laws, regulations, and enforcement practices are evolving and may have a material adverse effect on our results of operations, cash flows and financial position. Tax laws, regulations, and administrative practices in various jurisdictions are evolving and may be subject to significant changes due to economic, political, and other conditions. There are many transactions that occur during the ordinary course of business for which the ultimate tax determination is uncertain, and significant judgment is required in evaluating and estimating our provision and accruals for taxes. Governments are increasingly focused on ways to increase tax revenues, particularly from multinational corporations, which may lead to an increase in audit activity and aggressive positions taken by tax authorities.
Changes or clarifications to U.S. tax laws could materially affect the tax treatment of our domestic and foreign earnings. The Organisation for Economic Co-operation and Development, an international association of 34 countries, including the United States, released the final reports from its Base Erosion and Profit Shifting, or BEPS, Action Plans, which aim to standardize and modernize global tax policies. The BEPS Action Plans propose revisions to numerous tax rules, including country-by-country reporting, permanent establishment, hybrid entities and instruments, transfer pricing, and tax treaties. The BEPS Action Plans have been or are being enacted by countries where we have operations.
Developments in relevant tax laws, regulations, administrative practices and enforcement practices could have a material adverse effect on our operating results, financial position and cash flows, including the need to obtain additional financing.
We are subject to tax audits by various tax authorities in many jurisdictions. Our income tax returns are based on calculations and assumptions that require significant judgment, and are subject to audit by various tax authorities. In addition, the calculation of our tax liabilities involves dealing with uncertainties in the application of complex tax laws. We regularly assess the potential outcomes of examinations by tax authorities in determining the adequacy of our provision for income taxes.
In connection with the audit by the Australian Taxation Office, or ATO, for the tax years 2009 to 2013, we received Notices of Amended Assessments in March 2018. Based on these assessments, the ATO asserted that we owe $151.7 million in additional income tax and $38.4 million in accrued interest. We agreed to a payment arrangement with the ATO, whereby an amount of $75.9 million was paid by us in April 2018, with the remaining amounts due only if we are unsuccessful in defending our position. In June 2018, we received a notice from the ATO claiming penalties of 50% of the additional income tax that was assessed or $75.9 million. In accordance with the payment arrangement, all remaining tax, interest and penalty amounts outstanding are due only if we are unsuccessful in defending our position. We do not agree with the ATO’s assessments and intend to pursue administrative and legal steps to defend our position. We continue to believe we are more likely than not to be successful in defending our position. However, if we are not successful, there may be material changes to our past or future taxable income, tax payable or deferred tax assets, we will not receive a refund of the $75.9 million we paid in April 2018, and we will be required to pay penalties and interest that could materially adversely affect our financial results. The ATO is currently auditing tax years 2014 to 2018 and may advance the position that additional taxes are owed for those years as well.
Our quarterly operating results are subject to fluctuation for a variety of reasons. Our operating results have, from time to time, fluctuated on a quarterly basis and may be subject to similar fluctuations in the future. These fluctuations may result from a number of factors, including:
the introduction of new products by us or our competitors;
the geographic mix of product sales;
the success and costs of our marketing efforts in new regions;
changes in third-party payor reimbursement;
timing of regulatory clearances and approvals;
costs associated with acquiring and integrating new businesses, technologies and product offerings;
timing of orders by distributors;
expenditures incurred for research and development;
competitive pricing in different regions;
the effect of foreign currency transaction gains or losses; and
other activities of our competitors.
Fluctuations in our quarterly operating results may cause the market price of our common stock to fluctuate.
If a natural or man-made disaster strikes our manufacturing facilities, we will be unable to manufacture our products for a substantial amount of time and our sales and profitability will decline. Our facilities and the manufacturing equipment we use to produce our products would be costly to replace and could require substantial lead-time to repair or replace. The facilities may be affected by natural or man-made disasters, including COVID-19 that has spread globally, and in the event they were affected by a disaster, we would be forced to rely on third-party manufacturers. Although we believe we possess adequate insurance for the disruption of our business from causalities, such insurance may not be sufficient to cover all of our potential losses and may not continue to be available to us on acceptable terms, or at all.
Delaware law and provisions in our charter and could make it difficult for another company to acquire us. Provisions of our certificate of incorporation may have the effect of delaying or preventing changes in control or management which might be beneficial to us or our security holders. In particular, our board of directors has the authority to issue up to 2,000,000 shares of preferred stock and to determine the price, rights, preferences, privileges and restrictions, including voting rights, of those shares without further vote or action by the stockholders. The rights of the holders of our common stock will be subject to, and may be adversely affected by, the rights of the holders of any preferred stock that may be issued in the future. The issuance of preferred stock may have the effect of delaying, deferring or preventing a change in control, may discourage bids for our common stock at a premium over the market price of our common stock and may adversely affect the market price of our common stock and the voting and other rights of the holders of our common stock.
You may not be able to enforce the judgments of U.S. courts against some of our assets or officers and directors. A substantial portion of our assets are located outside the United States. Additionally, some of our directors and executive officers reside outside the United States, along with all or a substantial portion of their assets. As a result, it may not be possible for investors to enforce judgments of U.S. courts relating to any liabilities under U.S. securities laws against our assets, those persons or their assets. In addition, investors may not be able to pursue claims based on U.S. securities laws against these assets or these persons in non-U.S. courts, where most of these assets and persons reside.
We are increasingly dependent on information technology systems and infrastructure. Our technology systems are potentially vulnerable to breakdown or other interruption by fire, power loss, system malfunction, unauthorized access and other events. Likewise, data privacy breaches by employees and others with both permitted and unauthorized access to our systems may pose a risk that sensitive data may be exposed to unauthorized persons or to the public, or may be permanently lost. While we have invested heavily in the protection of data and information technology and in related training, there can be no assurance that our efforts will prevent significant breakdowns, breaches in our systems or other cyber incidents that could have a material adverse effect upon the reputation, business, operations or financial condition of the company. In addition, significant implementation issues may arise as we continue to consolidate and outsource certain computer operations and application support activities.
Our results of operations may be materially affected by global economic conditions generally, including conditions in the financial markets. Recently, concerns over inflation, energy costs, geopolitical issues, the availability and cost of credit, the impact of the COVID-19 pandemic, and the ability of sovereign nations to pay their debts have contributed to increased volatility and diminished expectations for the economy and the financial markets going forward. These factors, combined with volatile commodity prices, declining business and consumer confidence and increased unemployment, have precipitated an economic slowdown. It is difficult to predict how long the current economic conditions will continue and whether the economic conditions will continue to deteriorate. If the economic climate in the United States or outside the United States continues to deteriorate or there is a shift in government spending priorities, customers or potential customers could reduce or delay their purchases, which could impact our revenue, our ability to manage inventory levels, collect customer receivables, and ultimately decrease our profitability.
Our leverage and debt service obligations could adversely affect our business. As of June 30, 2020, our total consolidated debt was $1.2 billion. We may incur additional indebtedness in the future. Our indebtedness could have adverse consequences, including:
making it more difficult to satisfy our financial obligations;
increasing our vulnerability to adverse economic, regulatory and industry conditions;
limiting our ability to compete and our flexibility in planning for, or reacting to, changes in our business and the industry in which we operate;
limiting our ability to borrow additional funds for working capital, capital expenditure, acquisitions and general corporate or other purposes; and
exposing us to greater interest rate risk.
Our debt service obligations will require us to use a portion of our operating cash flow to pay interest and principal in indebtedness, which could impede our growth. Our ability to make payments on, and to refinance, our indebtedness, and to fund capital expenditures will depend on our ability to generate cash in the future. This is subject to general economic, financial, competitive, legislative, regulatory, and other factors, many of which are beyond our control.
We may be adversely affected by recent proposals to reform LIBOR. Certain of our financial arrangements, including credit facilities, are made at variable interest rates that use the London Interbank Offered Rate, or LIBOR (or metrics derived from or related to LIBOR), as a benchmark for establishing the interest rate. On July 27, 2017, the United Kingdom’s Financial Conduct Authority announced that it intends to stop persuading or compelling banks to submit LIBOR rates after 2021. These reforms may cause LIBOR to cease to exist, new methods of calculating LIBOR to be established, or alternative reference rates to be established. The Alternative Reference Rates Committee (ARRC) has proposed that the Secured Overnight Financing Rate (SOFR) is the rate that represents best practice as the alternative to LIBOR for use in financial and other derivatives contracts that are currently indexed to United States dollar LIBOR. ARRC has proposed a paced market transition plan to SOFR from LIBOR, and organizations are currently working on industry wide and company specific transition plans as it relates to financial and other derivative contracts exposed to LIBOR. Uncertainty exists as to the transition process and broad acceptance of SOFR as the primary alternative to LIBOR, and the potential consequences to us cannot be fully predicted. Changes in market interest rates may influence our financing costs, returns on financial investments and the valuation of derivative contracts and could reduce our earnings and cash flows.
We may impair intangible assets, such as goodwill. We have recorded intangible assets, including goodwill in connection with our acquisitions. At least on an annual basis, we will evaluate whether facts and circumstances indicate any impairment of the values of these intangible assets. As circumstances change, we cannot assure you that the value of these intangible assets will be realized by us. If we determine that a significant impairment has occurred, we will be required to write-off the impaired portion of intangible assets, which could have a material adverse effect on our results of operations in the period in which the write-off occurs.
ITEM 1B UNRESOLVED STAFF COMMENTS
We have received no written comments regarding our periodic or current reports from the staff of the SEC that were issued 180 days or more before the end of our fiscal year 2020 that remain unresolved.
ITEM 2 PROPERTIES
We conduct our operations in both owned and leased properties. Our principal executive offices and U.S. sales facilities consist of approximately 230,000 square feet and are located on Spectrum Center Boulevard in San Diego, California, in a building we own. We have our primary research and development facilities, as well as office and manufacturing facilities at our owned site in Sydney, Australia. Other facilities are leased in Atlanta, Georgia, and Moreno Valley, California, U.S.A.; Loyang and Galaxis, Singapore; Munich, Germany; Lyon, France; Suzhou, China; and Johor Bahru, Malaysia. We are establishing a new manufacturing facility in Tuas, Singapore that will eventually replace our Loyang facility.
We believe that our facilities are adequate to meet the needs of our current business operations. At June 30, 2020, our principal owned and leased properties were as follows:
|
|
|
|
|
|
Location |
Ownership Status |
Square |
|
Primary Usage |
|
|
San Diego, California |
Owned |
230,000 |
|
Corporate headquarters, sales and administration |
|
Sydney, Australia |
Owned |
224,000 |
|
Manufacturing, engineering, research and development, sales and administration |
|
Suzhou, China |
Owned |
53,000 |
|
Manufacturing, engineering, research and development |
|
Atlanta, Georgia |
Leased |
522,000 |
|
Warehouse and distribution; SaaS sales and administration, engineering, research and development |
|
Moreno Valley, California |
Leased |
244,000 |
|
Warehouse and distribution |
|
Tuas, Singapore |
Leased |
268,000 |
|
Future manufacturing facility, currently being established |
|
Munich, Germany |
Leased |
109,000 |
|
Sales and distribution |
|
Loyang, Singapore |
Leased |
95,000 |
|
Manufacturing facility, engineering, research and development |
|
Minneapolis, United States |
Leased |
86,000 |
|
SaaS sales and administration, engineering, research and development |
|
Chatsworth, California |
Leased |
72,000 |
|
Motor manufacturing, engineering, research and development |
|
Lyon, France |
Leased |
52,000 |
|
Sales and distribution |
|
Halifax, Canada |
Leased |
47,000 |
|
Engineering, research and development |
|
Johor Bahru, Malaysia |
Leased |
46,000 |
|
Engineering, research and development |
ITEM 3 LEGAL PROCEEDINGS
We are involved in various legal proceedings, claims, investigations and litigation that arise in the ordinary course of our business. We investigate these matters as they arise, and accrue estimates for resolution of legal and other contingencies in accordance with Statement of Financial Accounting Standard No. 5. See Note 17 – Legal Actions, Contingencies and Commitments of the Notes to Consolidated Financial Statements (Part II, Item 8) included in this report.
Litigation is inherently uncertain. Accordingly, we cannot predict with certainty the outcome of these matters. But we do not expect the outcome of these matters to have a material adverse effect on our consolidated financial statements when taken as a whole.
ITEM 4 MINE SAFETY DISCLOSURES
Not Applicable.
PART II
ITEM 5 MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Our common stock is traded on the NYSE under the symbol “RMD”. As of July 31, 2020, there were 26 holders of record of our common stock, although the actual number of stockholders of our common stock is greater than this number of holders of record and many of these holders of record own shares as nominees on behalf of other beneficial owners.
Securities Authorized for Issuance Under Equity Compensation Plans
The information included under Item 12 of Part III of this Report, “Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters,” is hereby incorporated by reference into this Item 5 of Part II of this Report.
Purchases of Equity Securities
On February 21, 2014, our board of directors approved our current share repurchase program, authorizing us to acquire up to an aggregate of 20.0 million shares of our common stock. The program allows us to repurchase shares of our common stock from time to time for cash in the open market, or in negotiated or block transactions, as market and business conditions warrant and subject to applicable legal requirements. There is no expiration date for this program, and the program may be accelerated, suspended, delayed or discontinued at any time at the discretion of our board of directors. All share repurchases after February 21, 2014 have been executed under this program.
In fiscal year 2019, we temporarily suspended our share repurchase program due to recent acquisitions. As a result, we did not repurchase any shares during the twelve months ended June 30, 2020. However, there is no expiration date for this program, and we may, at any time, elect to resume the share repurchase program as the circumstances allow. Since the inception of the share buyback programs, we have repurchased 41.8 million shares at a total cost of $1.6 billion. At June 30, 2020, 12.9 million additional shares can be repurchased under the approved share repurchase program.
PERFORMANCE GRAPH
This performance graph is furnished and shall not be deemed “filed” with the SEC or subject to Section 18 of the Exchange Act, nor shall it be deemed incorporated by reference in any of our filings under the Securities Act of 1933, as amended.
The following graph compares the cumulative total stockholders return on our common stock from June 30, 2015 through June 30, 2020, with the comparable cumulative return of the S&P 500 index, the S&P 500 Health Care index, and the Dow Jones U.S. Medical Devices index. The graph assumes that $100 was invested in our common stock and each index on June 30, 2015. In addition, the graph assumes the reinvestment of all dividends paid. The stock price performance on the following graph is not necessarily indicative of future stock price performance.
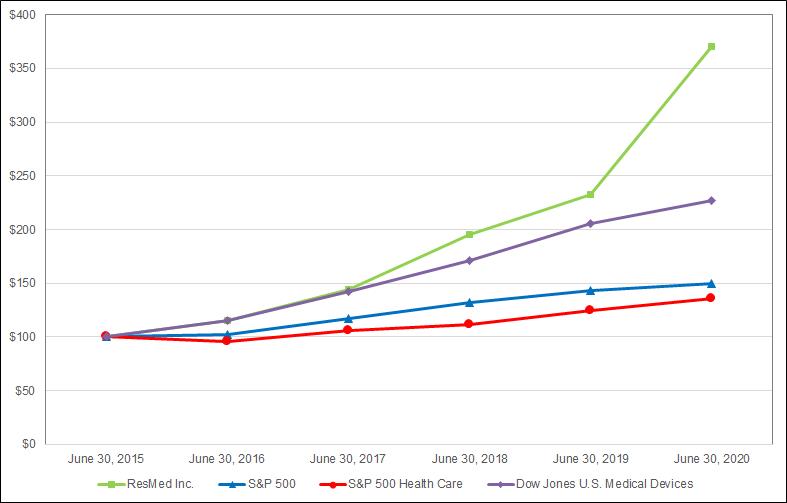
The following table shows total indexed return of stock price plus reinvestments of dividends, assuming an initial investment of $100 at June 30, 2015, for the indicated periods.
|
|
|
|
|
|
|
|
As of June 30, |
|||||
Index |
2015 |
2016 |
2017 |
2018 |
2019 |
2020 |
ResMed Inc. |
100 |
115 |
144 |
195 |
233 |
370 |
S&P 500 |
100 |
102 |
117 |
132 |
143 |
150 |
S&P 500 Health Care |
100 |
96 |
106 |
112 |
125 |
136 |
Dow Jones U.S. Medical Devices |
100 |
115 |
142 |
171 |
206 |
227 |
ITEM 6 SELECTED FINANCIAL DATA
The following table summarizes certain selected consolidated financial data for, and as of the end of, each of the fiscal years in the five-year period ended June 30, 2020. The data set forth below should be read together with Item 7 of Part II of this report, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and Item 8 of Part II of this report, “Consolidated Financial Statements and Supplementary Data”, and related notes included elsewhere in this report. The consolidated statement of income data for the years ended June 30, 2020, 2019 and 2018 and the consolidated balance sheet data as of June 30, 2020 and 2019 are derived from our audited consolidated financial statements included elsewhere in this report. The consolidated statement of income data for the years ended June 30, 2017 and 2016 and the consolidated balance sheet data as of June 30, 2018, 2017 and 2016 are derived from our audited consolidated financial statements not included in this report. Historical results do not necessarily indicate the results to be expected in the future, and the results for the years presented should not be considered to indicate our future results of operations.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Consolidated Statement of Income Data |
|
Years Ended June 30, |
|||||||||||||
(In thousands, except per share data): |
|
2020 |
|
2019 |
|
2018 |
|
2017 |
|
2016 |
|||||
Net revenue |
|
|
2,957,013 |
|
|
2,606,572 |
|
|
2,340,196 |
|
|
2,066,737 |
|
|
1,838,713 |
Cost of sales (exclusive of amortization shown separately below) |
|
|
1,189,624 |
|
|
1,069,987 |
|
|
978,032 |
|
|
864,992 |
|
|
772,216 |
Amortization of acquired intangible assets* |
|
|
49,603 |
|
|
42,514 |
|
|
27,266 |
|
|
29,477 |
|
|
12,906 |
Total cost of sales |
|
|
1,239,227 |
|
|
1,112,501 |
|
|
1,005,298 |
|
|
894,469 |
|
|
785,122 |
Gross profit |
|
|
1,717,786 |
|
|
1,494,071 |
|
|
1,334,898 |
|
|
1,172,268 |
|
|
1,053,591 |
Selling, general and administrative expenses |
|
|
676,689 |
|
|
645,010 |
|
|
600,369 |
|
|
553,968 |
|
|
488,057 |
Research and development expenses |
|
|
201,946 |
|
|
180,651 |
|
|
155,149 |
|
|
144,467 |
|
|
118,651 |
Amortization of acquired intangible assets* |
|
|
30,092 |
|
|
32,424 |
|
|
19,117 |
|
|
17,101 |
|
|
11,017 |
Restructuring expenses |
|
|
- |
|
|
9,401 |
|
|
18,432 |
|
|
12,358 |
|
|
6,914 |
Litigation settlement expenses |
|
|
(600) |
|
|
41,199 |
|
|
- |
|
|
8,500 |
|
|
- |
Acquisition related expenses |
|
|
- |
|
|
6,123 |
|
|
- |
|
|
10,076 |
|
|
- |
Total operating expenses |
|
|
908,127 |
|
|
914,808 |
|
|
793,067 |
|
|
746,470 |
|
|
624,639 |
Income from operations |
|
|
809,659 |
|
|
579,263 |
|
|
541,831 |
|
|
425,798 |
|
|
428,952 |
Other income: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Interest income (expense), net |
|
|
(39,356) |
|
|
(33,857) |
|
|
(11,977) |
|
|
(11,151) |
|
|
5,654 |
Loss attributable to equity method investments |
|
|
(25,058) |
|
|
(15,833) |
|
|
- |
|
|
- |
|
|
- |
Other, net |
|
|
(12,157) |
|
|
(10,726) |
|
|
(8,542) |
|
|
4,096 |
|
|
4,960 |
Total other income (loss), net |
|
|
(76,571) |
|
|
(60,416) |
|
|
(20,519) |
|
|
(7,055) |
|
|
10,614 |
Income before income taxes |
|
|
733,088 |
|
|
518,847 |
|
|
521,312 |
|
|
418,743 |
|
|
439,566 |
Income taxes |
|
|
111,414 |
|
|
114,255 |
|
|
205,724 |
|
|
76,459 |
|
|
87,157 |
Net income |
|
$ |
621,674 |
|
$ |
404,592 |
|
$ |
315,588 |
|
$ |
342,284 |
|
$ |
352,409 |
Basic earnings per share |
|
$ |
4.31 |
|
$ |
2.83 |
|
$ |
2.21 |
|
$ |
2.42 |
|
$ |
2.51 |
Diluted earnings per share |
|
$ |
4.27 |
|
$ |
2.80 |
|
$ |
2.19 |
|
$ |
2.40 |
|
$ |
2.49 |
Dividends per share |
|
$ |
1.56 |
|
$ |
1.48 |
|
$ |
1.40 |
|
$ |
1.32 |
|
$ |
1.20 |
Weighted average: |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Basic shares outstanding |
|
|
144,338 |
|
|
143,111 |
|
|
142,764 |
|
|
141,360 |
|
|
140,242 |
Diluted shares outstanding |
|
|
145,652 |
|
|
144,484 |
|
|
143,987 |
|
|
142,453 |
|
|
141,669 |
* Within our consolidated statements of income for the years ended June 30, 2020, 2019, 2018, 2017 and 2016, cost of sales has been adjusted to include amortization of acquired intangible assets directly applicable to revenue. As a result, gross profit includes amortization of acquired intangible assets relating to cost of sales and operating expenses have been reduced by this amount. There was no impact on income from operations, income before taxes or net income, as a result of this reclassification.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
As of June 30, |
|||||||||||||
Consolidated Balance Sheet Data (In thousands): |
|
2020 |
|
2019 |
|
2018 |
|
2017 |
|
2016 |
|||||
Working capital |
|
$ |
920,698 |
|
$ |
589,375 |
|
$ |
554,468 |
|
$ |
1,283,877 |
|
$ |
781,730 |
Total assets |
|
|
4,587,376 |
|
|
4,107,682 |
|
|
3,063,923 |
|
|
3,468,487 |
|
|
3,256,705 |
Long-term debt, less current maturities |
|
|
1,164,133 |
|
|
1,258,861 |
|
|
269,988 |
|
|
1,078,611 |
|
|
873,332 |
Total stockholders’ equity |
|
$ |
2,497,027 |
|
$ |
2,072,193 |
|
$ |
2,058,980 |
|
$ |
1,960,266 |
|
$ |
1,694,831 |
Table of Contents
PART II |
Item 7 |
RESMED INC. AND SUBSIDIARIES
Management’s Discussion and Analysis of Financial Condition and Results of Operations
ITEM 7MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Overview
Management’s discussion and analysis of financial condition and results of operations is intended to help the reader understand the results of operations and financial condition of ResMed Inc. and subsidiaries. It is provided as a supplement to, and should be read together with the selected financial data and consolidated financial statements and notes included elsewhere in this report.
We are a global leader in the development, manufacturing, distribution and marketing of medical devices and cloud-based software applications that diagnose, treat and manage respiratory disorders, including sleep apnea, COPD, neuromuscular disease and other chronic diseases. Sleep apnea includes obstructive sleep apnea and other respiratory disorders that occur during sleep. Our products and solutions are designed to improve patient quality of life, reduce the impact of chronic disease and lower healthcare costs as global healthcare systems continue to drive a shift in care from hospitals to the home and lower cost settings. Our cloud-based digital health applications, along with our devices, are designed to provide connected care to improve patient outcomes and efficiencies for our customers.
Since the development of continuous positive airway pressure therapy, we have expanded our business by developing or acquiring a number of products and solutions for a broader range of respiratory disorders including technologies to be applied in medical and consumer products, ventilation devices, diagnostic products, mask systems, headgear and other accessories, dental devices, portable oxygen concentrators and cloud-based software informatics solutions to manage patient outcomes and customer and provider business processes. Our growth has been fueled by geographic expansion, our research and product development efforts, acquisitions and an increasing awareness of sleep apnea and other respiratory conditions like chronic obstructive pulmonary disease as significant health concerns.
We are committed to ongoing investment in research and development and product enhancements. During fiscal year 2020, we invested $201.9 million on research and development activities, which represents 6.8% of net revenues with a continued focus on the development and commercialization of new, innovative products and solutions that improve patient outcomes, create efficiencies for our customers and help physicians and providers better manage chronic disease and lower healthcare costs. During fiscal year 2020, we released new products including AirFit N30, a nasal cradle mask with a front-facing tube, and AirFit F30i, a top-of-head connected full face mask as well as expanded our AirView offering to include certain respiratory care devices. Due to multiple acquisitions, including of Brightree in April 2016, HEALTHCAREfirst in July 2018 and MatrixCare in November 2018, our operations now include out-of-hospital software platforms designed to support the professionals and caregivers who help people stay healthy in the home or care setting of their choice. These platforms comprise our SaaS business. These products, our cloud-based remote monitoring and therapy management system, and a robust product pipeline, should continue to provide us with a strong platform for future growth.
We have determined that we have two operating segments, which are the sleep and respiratory disorders sector of the medical device industry (“Sleep and Respiratory Care”) and the supply of business management software as a service to out-of-hospital health providers (“SaaS”).
Net revenue in fiscal year 2020 increased to $2,957.0 million, an increase of 13% compared to fiscal year 2019. Gross profit increased for the year ended June 30, 2020 to $1,717.8 million, from $1,494.1 million for the year ended June 30, 2019, an increase $223.7 million or 15%. Our net income for the year ended June 30, 2020 was $621.7 million or $4.27 per diluted share compared to net income of $404.6 million or $2.80 per diluted share for the year ended June 30, 2019.
Total operating cash flow for fiscal year 2020 was $802.3 million and at June 30, 2020, our cash and cash equivalents totaled $463.2 million. At June 30, 2020, our total assets were $4.6 billion and our stockholders’ equity was $2.5 billion. We paid a quarterly dividend of $0.39 per share during fiscal 2020 with a total amount of $225.1 million paid to stockholders.
In order to provide a framework for assessing how our underlying businesses performed, excluding the effect of foreign currency fluctuations, we provide certain financial information on a “constant currency basis”, which is in addition to the actual financial information presented. In order to calculate our constant currency information, we translate the current period financial information using the foreign currency exchange rates that were in effect during the previous comparable period. However, constant currency measures should not be considered in isolation or as an alternative to U.S. dollar measures that reflect current period exchange rates, or to other financial measures calculated and presented in accordance with U.S. generally accepted accounting principles.
For discussion related to the results of operations and changes in financial condition for the fiscal year ended June 30, 2019 compared to fiscal year June 30, 2018, please refer to Item 7 of Part II, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our Annual Report for the Year Ended June 30, 2019, which was filed with the United States Securities and Exchange Commission on August 18, 2019.
Table of Contents
PART II |
Item 7 |
RESMED INC. AND SUBSIDIARIES
Management’s Discussion and Analysis of Financial Condition and Results of Operations
Impact of COVID-19
In March 2020, the World Health Organization declared the outbreak of a novel strain of coronavirus (“COVID-19”) as a pandemic. Our primary goal during the COVID-19 pandemic is the preservation of life. We have prioritized protecting the health and safety of our employees and continuing to use our employees’ talents and our resources to help society meet and overcome the challenges the pandemic poses.
We have observed increased demand for our ventilator devices and masks, which can be used to treat COVID-19 patients. Due to governments’ varying restrictions on international and domestic travel, access to labor for our manufacturing facilities was impacted as was the availability of raw materials and components, which constrained our manufacturing capacity and restricted our ability to initially meet the substantial demand for ventilators. Our primary focus is maximizing the availability of our ventilators and other respiratory support devices for the patients that need them the most in the countries facing the greatest challenges. The global increase in our sales for these respiratory care products during fiscal year 2020 generally followed infection patterns around the world. We believe the global demand for these devices has largely been met, however, this may change depending on the ability for regions to contain and control infection rates, which remains highly uncertain. Additionally, as more becomes known about the virus and as governments pursue testing and vaccines, we may see an overall reduction in demand, and then face a corresponding risk of oversupply by us and by our competitors. While further outbreaks in the future are highly uncertain, we expect lower demand for ventilator products for the fiscal year ending June 30, 2021.
As anticipated, we observed lower demand for our sleep devices and masks during the three months ended June 30, 2020, and we continue to expect COVID-19 will lead to a temporary decrease in demand for these products from new patients for some or all of our fiscal year 2021. Specifically, diagnostic pathways for sleep apnea treatment, including HME suppliers and sleep clinics, have been impacted and, in some instances, been required, or in the future may be required, to temporarily close due to governments’ “shelter-in-place” orders, quarantines or similar orders or restrictions enacted to control the spread of COVID-19. In some countries, new patients are prescribed sleep apnea treatment through hospitals that are directing their resources to critical care, including COVID-19 treatment. The impact on these diagnostic and prescription pathways has likely resulted in a decrease in demand from new patients for our products designed to treat sleep apnea. Given the ongoing uncertainty regarding the duration and extent of the COVID-19 pandemic and measures taken to control the spread of COVID-19, we are uncertain as to the duration and extent of decreased demand for our sleep devices. However, due to the nature of the installed base of existing patients using our devices, we expect the demand for re-supply of our masks to be less impacted compared to devices.
Our SaaS business may also be affected by COVID-19 and measures taken to control the spread of COVID-19. Some of our existing and potential SaaS customers are HME distributors and, therefore, have been impacted, or may be impacted, by the same temporary business closures noted above. We also have existing and potential SaaS customers that operate care facilities and are either receiving and treating patients infected with COVID-19 or are implementing significant measures to safeguard their facilities against a potential COVID-19 outbreak. Given these challenging business conditions and the uncertain economic environment, we expect businesses will be deterred from adopting new or changing SaaS platforms, which may adversely impact our ability to engage new customers for our SaaS businesses, or expand the services used by existing customers.
Our ability to continue to operate without any significant negative impacts will in part depend on our ability to protect our employees. We have endeavored and continue to follow recommended actions of government and health authorities to protect our employees worldwide, but since COVID-19 was declared a pandemic in March 2020, we were able to broadly maintain our operations, and we are beginning the slow and careful process of progressively returning to work in our offices around the world. The pandemic has not negatively impacted our liquidity position.
Table of Contents
PART II |
Item 7 |
RESMED INC. AND SUBSIDIARIES
Management’s Discussion and Analysis of Financial Condition and Results of Operations
Fiscal Year Ended June 30, 2020 Compared to Fiscal Year Ended June 30, 2019
Net Revenues. Net revenue for the year ended June 30, 2020 increased to $2,957.0 million from $2,606.6 million for the year ended June 30, 2019, an increase of $350.4 million or 13% (a 15% increase on a constant currency basis). The following table summarizes our net revenue disaggregated by segment, product and region for the year ended June 30, 2020 compared to the year ended June 30, 2019 (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Year Ended June 30, |
||||||||||
|
|
2020 |
|
2019 |
|
% Change |
|
|
Constant Currency* |
|||
U.S., Canada and Latin America |
|
|
|
|
|
|
|
|
|
|
|
|
Devices |
|
$ |
792,766 |
|
$ |
743,066 |
|
7 |
% |
|
|
|
Masks and other |
|
|
779,561 |
|
|
677,430 |
|
15 |
|
|
|
|
Total Sleep and Respiratory Care |
|
$ |
1,572,327 |
|
$ |
1,420,496 |
|
11 |
|
|
|
|
Software as a Service |
|
|
354,632 |
|
|
275,789 |
|
29 |
|
|
|
|
Total |
|
$ |
1,926,959 |
|
$ |
1,696,285 |
|
14 |
|
|
|
|
Combined Europe, Asia and other markets |
|
|
|
|
|
|
|
|
|
|
|
|
Devices |
|
$ |
715,056 |
|
$ |
618,525 |
|
16 |
% |
|
19 |
% |
Masks and other |
|
|
314,998 |
|
|
291,762 |
|
8 |
|
|
11 |
|
Total Sleep and Respiratory Care |
|
$ |
1,030,054 |
|
$ |
910,287 |
|
13 |
|
|
16 |
|
Global revenue |
|
|
|
|
|
|
|
|
|
|
|
|
Devices |
|
$ |
1,507,822 |
|
$ |
1,361,591 |
|
11 |
% |
|
12 |
% |
Masks and other |
|
|
1,094,559 |
|
|
969,192 |
|
13 |
|
|
14 |
|
Total Sleep and Respiratory Care |
|
$ |
2,602,381 |
|
$ |
2,330,783 |
|
12 |
|
|
13 |
|
Software as a Service |
|
|
354,632 |
|
|
275,789 |
|
29 |
|
|
29 |
|
Total |
|
$ |
2,957,013 |
|
$ |
2,606,572 |
|
13 |
|
|
15 |
|
*Constant currency numbers exclude the impact of movements in international currencies.
Sleep and Respiratory Care
Net revenue from our Sleep and Respiratory Care business for the year ended June 30, 2020 increased to $2,602.4 million from $2,330.8 million for the year ended June 30, 2019, an increase of $271.6 million or 12%. Movements in international currencies against the U.S. dollar negatively impacted net revenues by approximately $29.9 million for the year ended June 30, 2020. Excluding the impact of currency movements, total net revenue from our Sleep and Respiratory Care business for the year ended June 30, 2020 increased by 13% compared to the year ended June 30, 2019. The increase in net revenue was primarily attributable to an increase in unit sales of our devices and masks, including as a result of increased demand for our ventilators due to COVID-19.
Net revenue from our Sleep and Respiratory Care business in the United States, Canada and Latin America for the year ended June 30, 2020 increased to $1,572.3 million from $1,420.5 million for the year ended June 30, 2019, an increase of $151.8 million or 11%. The increase was primarily due to an increase in unit sales of our devices and masks, including as a result of increased demand for our ventilators due to COVID-19.
Net revenue from our Sleep and Respiratory Care business in markets in combined Europe, Asia and other markets increased for the year ended June 30, 2020 to $1,030.1 million from $910.3 million for the year ended June 30, 2019, an increase of $119.8 million or 13% (an increase of 16% on a constant currency basis). The constant currency increase in sales in combined Europe, Asia and other markets predominantly reflects an increase in unit sales of our devices and masks, including as a result of increased demand for our ventilators due to COVID-19.
Net revenue from devices for the year ended June 30, 2020 increased to $1,507.8 million from $1,361.6 million for the year ended June 30, 2019, an increase of $146.2 million or 11%, including an increase of 7% in the United States, Canada and Latin America and an increase of 16% in combined Europe, Asia and other markets (a 19% increase on a constant currency basis). Excluding the impact of foreign currency movements, device sales for the year ended June 30, 2020 increased by 12%.
Net revenue from masks and other for the year ended June 30, 2020 increased to $1,094.6 million from $969.2 million for the year ended June 30, 2019, an increase of 13%, including an increase of 15% in the United States, Canada and Latin America and an increase of 8% in combined Europe, Asia and other markets (an 11% increase on a constant currency basis). Excluding the impact of foreign currency movements, masks and other sales increased by 14%, compared to the year ended June 30, 2019.
Table of Contents
PART II |
Item 7 |
RESMED INC. AND SUBSIDIARIES
Management’s Discussion and Analysis of Financial Condition and Results of Operations
Software as a Service
Net revenue from our SaaS business for the year ended June 30, 2020 was $354.6 million, compared to $275.8 million for the year ended June 30, 2019, an increase of $78.8 million or 29%. The increase was predominantly due to revenue attributable to MatrixCare, which was acquired on November 13, 2018, and continued growth in our SaaS product offerings.
Gross Profit and Gross Margin. Within our consolidated statements of income for the years ended June 30, 2020, 2019 and 2018, cost of sales has been adjusted to include amortization of acquired intangible assets directly applicable to revenue. As a result, gross profit now includes amortization of acquired intangible assets relating to cost of sales and operating expenses have been reduced by this amount. There was no impact on income from operations, income before taxes or net income, as a result of this reclassification. The adjustments to the previously reported amounts are not material.
The table below presents a reconciliation of amortization of acquired intangible assets by income statement caption summing to total amortization of acquired intangible assets as previously reported for the year ended June 30, 2019 (in thousands):
|
|
|
|
|
|
2019 |
|
Amortization of acquired intangible assets related to cost of sales |
|
$ |
42,514 |
Amortization of acquired intangible assets related to operating expenses |
|
|
32,424 |
Total as previously reported |
|
$ |
74,938 |
The table below presents a reconciliation of gross profit as previously reported for the year ended June 30, 2019 adjusted for the amortization of acquired intangible assets now included in cost of sales (in thousands):
|
|
|
|
|
|
2019 |
|
Gross profit as previously reported |
|
$ |
1,536,585 |
Amortization of acquired intangible assets related to cost of sales |
|
|
(42,514) |
Gross profit |
|
$ |
1,494,071 |
Gross profit increased for the year ended June 30, 2020 to $1,717.8 million from $1,494.1 million for the year ended June 30, 2019, an increase of $223.7 million or 15%. Gross profit as a percentage of net revenue was 58.1% for the year ended June 30, 2020, compared with the 57.3% for the year ended June 30, 2019. The increase in gross margin was due primarily to favorable product mix, which was partially offset by an increase in manufacturing and logistics costs as a result of the COVID-19 pandemic and an increase in amortization of intangible assets associated with MatrixCare and Propeller Health, which were acquired in November 2018 and January 2019, respectively.
Selling, General and Administrative Expenses. Selling, general and administrative expenses increased for the year ended June 30, 2020 to $676.7 million from $645.0 million for the year ended June 30, 2019, an increase of $31.7 million or 5%. The selling, general and administrative expenses, as reported in U.S. dollars, were favorably impacted by the movement of international currencies against the U.S. dollar, which decreased our expenses by approximately $15.1 million. Excluding the impact of foreign currency movements, selling, general and administrative expenses for the year ended June 30, 2020 increased by 7% compared to the year ended June 30, 2019. As a percentage of net revenue, selling, general and administrative expenses for the year ended June 30, 2020 improved to 22.9% compared to 24.7% for the year ended June 30, 2019.
The constant currency increase in selling, general and administrative expenses was primarily due to additional personnel to support our commercial activities and additional expenses associated with the consolidation of our acquisitions of MatrixCare and Propeller Health, partially offset by a decrease in legal costs and travel, marketing and consulting expenses, either as a direct or indirect result of the COVID-19 pandemic.
Research and Development Expenses. Research and development expenses increased for the year ended June 30, 2020 to $201.9 million from $180.7 million for the year ended June 30, 2019, an increase of $21.3 million or 12%. The research and development expenses were favorably impacted by the movement of international currencies against the U.S. dollar, which decreased our expenses by approximately $4.4 million, as reported in U.S. dollars. Excluding the impact of foreign currency movements, research and development expenses for the year ended June 30, 2020 increased by 14% compared to the year ended June 30, 2019. As a percentage of net revenue, research and development expenses were 6.8% for the year ended June 30, 2020 compared to 6.9% for the year ended June 30, 2019.
The constant currency increase in research and development expenses was primarily due to additional expenses associated with the consolidation of our acquisitions of MatrixCare and Propeller Health as well as additional personnel to facilitate development of new products and solutions.
Table of Contents
PART II |
Item 7 |
RESMED INC. AND SUBSIDIARIES
Management’s Discussion and Analysis of Financial Condition and Results of Operations
Amortization of Acquired Intangible Assets. Amortization of acquired intangible assets for the year ended June 30, 2020 totaled $30.1 million compared to $32.4 million for the year ended June 30, 2019. The decrease in amortization expense was attributable to our historical intangible assets becoming fully amortized during the fiscal year.
Restructuring Expenses. During the year ended June 30, 2020, we did not incur material restructuring expenses. During the year ended June 30, 2019, we incurred restructuring expenses of $9.4 million associated with the reorganization, rationalization and relocation of some of our research and development and SaaS operations including the closure of our German research and development site. We recorded the full amount of $9.4 million during the year ended June 30, 2019, within our operating expenses, which was separately disclosed as restructuring expenses. The restructuring expenses consisted primarily of severance payments to employees and contract exit costs associated with several impacted sites.
Acquisition Related Expenses. During the year ended June 30, 2020, we did not incur material acquisition related expenses. During the year ended June 30, 2019, we recognized acquisition related expenses of $6.1 million associated primarily with our acquisition of MatrixCare.
Litigation Settlement Expenses. During the year ended June 30, 2020, we did not incur material litigation settlement expenses. During the year ended June 30, 2019, we recognized litigation settlement expenses of $41.2 million on account of a tentative agreement with the Department of Justice to resolve an ongoing investigation by the government into certain of our product offerings. The final agreement, entered into in December 2019, included payment by us of $39.5 million, and additional fees and administrative costs raising the overall total to $41.2 million.
Total Other Income (Loss), Net. Total other income (loss), net for the year ended June 30, 2020 was a loss of $76.6 million, compared to a loss of $60.4 million for the year ended June 30, 2019. The change was due primarily to an increase in losses attributable to equity method investments for the year ended June 30, 2020 of $25.1 million, compared to $15.8 million for the year ended June 30, 2019. The losses attributable to equity method investments relate to our joint venture with Verily whereby we recognize our share of the joint venture’s losses. Additionally, interest expense increased to $40.4 million for the year ended June 30, 2020 compared to interest expense of $36.2 million for the year ended June 30, 2019.
Income Taxes. Our effective income tax rate decreased to 15.2% for the year ended June 30, 2020 from 22.0% for the year ended June 30, 2019. Our effective income tax rate was affected by the geographic mix of our earnings and windfall tax benefits related to the vesting or settlement of employee share-based awards. Our Singapore operations operate under certain tax holidays and tax incentive programs that will expire in whole or in part at various dates through June 30, 2030. As a result of the U.S. Tax Act, we treated all non-U.S. historical earnings as taxable during the year ended June 30, 2018. Therefore, future repatriation of cash held by our non-U.S. subsidiaries will generally not be subject to U.S. federal tax, if repatriated.
Finally, in connection with the audit by the Australian Tax Office (the “ATO”) for the tax years 2009 to 2013, we received Notices of Amended Assessments in March 2018. Based on these assessments, the ATO asserted that we owe $151.7 million in additional income tax and $38.4 million in accrued interest, of which $75.9 million was paid in April 2018 under a payment arrangement with the ATO. As of June 30, 2020, we have recorded a receivable in prepaid taxes and other non-current assets for the amount paid as we ultimately expect this will be refunded by the ATO. In June 2018, we received a notice from the ATO claiming penalties of 50% of the additional income tax that was assessed or $75.9 million. The ATO is currently auditing tax years 2014 to 2018. We do not agree with the ATO’s assessments and continue to believe we are more likely than not to be successful in defending our position.
Net Income and Earnings per Share. As a result of the factors above, our net income for the year ended June 30, 2020 was $621.7 million compared to net income of $404.6 million for the year ended June 30, 2019. Our earnings per diluted share for the year ended June 30, 2020 was $4.27 compared to $2.80 for the year ended June 30, 2019, an increase of 53%.
Liquidity and Capital Resources
As of June 30, 2020 and June 30, 2019, we had cash and cash equivalents of $463.2 million and $147.1 million, respectively. Working capital was $920.7 million and $589.4 million, at June 30, 2020 and June 30, 2019, respectively. As of June 30, 2020 we had $1.2 billion of borrowings under our revolving credit facility, term credit facility and senior notes as compared to $1.3 billion at June 30, 2019. In response to the uncertainty associated with the COVID-19 pandemic, we increased our cash and cash equivalents position during the year by drawing down from our revolving credit facility. As of June 30, 2020, we had $1.1 billion available for draw down under the revolving credit facility and a combined total of $1.5 billion in cash and available liquidity under the revolving credit facility.
Table of Contents
PART II |
Item 7 |
RESMED INC. AND SUBSIDIARIES
Management’s Discussion and Analysis of Financial Condition and Results of Operations
As of June 30, 2020 and June 30, 2019, our cash and cash equivalent balances held within the United States amounted to $158.8 million and $33.6 million, respectively. Our remaining cash and cash equivalent balances at June 30, 2020 and June 30, 2019, of $304.4 million and $113.5 million, respectively, were held by our non-U.S. subsidiaries. Our cash and cash equivalent balances are held at highly rated financial institutions.
We repatriated $400.0 million and $360.0 million to the United States during both the years ended June 30, 2020 and 2019, respectively, from earnings generated in each of those years. The amount of the current year foreign earnings that we have repatriated to the United States in the past has been determined, and the amount that we expect to repatriate during fiscal year 2021 will be determined, based on a variety of factors, including current year earnings of our foreign subsidiaries, foreign investment needs and the cash flow needs we have in the United States, such as for the repayment of debt, dividend distributions, and other domestic obligations.
During the year ended June 30, 2018, as a result of the U.S. Tax Act, we treated all non-U.S. historical earnings as taxable, which resulted in additional tax expense of $126.9 million which was payable over the proceeding eight years. Therefore, future repatriation of cash held by our non-U.S. subsidiaries will generally not be subject to U.S. federal tax if repatriated. On June 14, 2019, the U.S. Treasury Department issued final and temporary regulations relating to the repatriation of non-U.S. earnings. As a result, in the event our non-U.S. earnings had not been permanently reinvested, deferred taxes of approximately $194.4 million in U.S. federal deferred tax and $5.2 million in U.S. state deferred taxes would have been recognized in the consolidated financial statements.
Inventories at June 30, 2020 were $416.9 million, an increase of $67.3 or 19% over the balance at June 30, 2019 of $349.6 million. The increase in inventories was required primarily to support our revenue growth, including increased demand for our ventilators due to COVID-19.
Accounts receivable, net of allowance for doubtful accounts, at June 30, 2020 were $474.6 million, a decrease of $53.9 million or 10% over the June 30, 2019 accounts receivable balance of $528.5 million. Accounts receivable days’ sales outstanding of 65 days at June 30, 2020 decreased by 2 days compared to 67 days at June 30, 2019. Our allowance for doubtful accounts as a percentage of total accounts receivable at June 30, 2020 and 2019 was 5.7% and 4.5%, respectively.
Effective July 1, 2019, we adopted the Accounting Standards Update (“ASU”) No. 2016-02, “Leases” (Topic 842). As of June 30, 2020, and in accordance with the new guidance, we have recognized a right-of-use asset (“ROU”) of $118.3 million and a lease liability of $123.1 million on the balance sheet for all operating leases, other than those that meet the definition of a short-term lease.
During the year ended June 30, 2020, we generated cash of $802.3 million from operations compared to $459.1 million for the year ended June 30, 2019. The increase in cash generated from operations during the year ended June 30, 2020 was primarily due to the increase in operating profit and improvement in working capital, partially offset by higher inventory levels. Movements in foreign currency exchange rates during the year ended June 30, 2020 had the effect of increasing our cash and cash equivalents by $10.9 million, as reported in U.S. dollars.
During the year ended June 30, 2020, we paid $27.9 million associated with business acquisitions, net of cash acquired, compared to $951.4 million during the year ended June 30, 2019.
We have temporarily suspended our share repurchase program and, accordingly, did not repurchase any shares during year ended June 30, 2020. During the year ended June 30, 2019, we repurchased 200,000 shares at a cost of $22.8 million under our share repurchase program. During fiscal years 2020 and 2019, we also paid dividends totaling $225.1 million and $211.7 million, respectively.
Details of contractual obligations at June 30, 2020 are as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Payments Due by June 30, |
||||||||||||||||
|
|
Total |
|
2021 |
|
2022 |
|
2023 |
|
2024 |
|
2025 |
|
Thereafter |
|||||||
Debt |
|
$ |
1,180,000 |
|
$ |
12,000 |
|
$ |
12,000 |
|
$ |
12,000 |
|
$ |
644,000 |
|
$ |
- |
|
$ |
500,000 |
Interest on debt |
|
|
155,129 |
|
|
26,435 |
|
|
26,435 |
|
|
24,816 |
|
|
16,725 |
|
|
16,725 |
|
|
43,994 |
Operating leases |
|
|
131,382 |
|
|
25,963 |
|
|
19,038 |
|
|
15,906 |
|
|
13,407 |
|
|
10,684 |
|
|
46,384 |
Purchase obligations |
|
|
462,996 |
|
|
458,623 |
|
|
3,678 |
|
|
518 |
|
|
111 |
|
|
66 |
|
|
- |
Total |
|
$ |
1,929,507 |
|
$ |
523,021 |
|
$ |
61,151 |
|
$ |
53,240 |
|
$ |
674,243 |
|
$ |
27,475 |
|
$ |
590,378 |
Table of Contents
PART II |
Item 7 |
RESMED INC. AND SUBSIDIARIES
Management’s Discussion and Analysis of Financial Condition and Results of Operations
Details of other commercial commitments at June 30, 2020 are as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Amount of Commitment Expiration Per Period |
||||||||||||||||
|
|
Total |
|
2021 |
|
2022 |
|
2023 |
|
2024 |
|
2025 |
|
Thereafter |
|||||||
Standby letter of credit |
|
$ |
16,318 |
|
$ |
3,638 |
|
$ |
33 |
|
$ |
536 |
|
$ |
- |
|
$ |
- |
|
$ |
12,111 |
Guarantees* |
|
|
3,302 |
|
|
185 |
|
|
24 |
|
|
38 |
|
|
49 |
|
|
18 |
|
|
2,988 |
Total |
|
$ |
19,620 |
|
$ |
3,823 |
|
$ |
57 |
|
$ |
574 |
|
$ |
49 |
|
$ |
18 |
|
$ |
15,099 |
*These guarantees mainly relate to requirements under contractual obligations with insurance companies transacting with our German subsidiaries and guarantees provided under our facility leasing obligations.
Refer to Note 17 - Legal Actions, Contingencies and Commitments of the Notes to the Consolidated Financial Statements (Part II, Item 8) for details of our contingent obligations under recourse provisions.
Segment Information
We have determined that we have two operating segments, which are the Sleep and Respiratory Care segment and the SaaS segment. See Note 15 – Segment Information of the Notes to the Consolidated Financial Statements (Part II, Item 8) for financial information regarding segment reporting. Financial information about our revenues from and assets located in foreign countries is also included in the notes to the consolidated financial statements included in this report.
Credit Facility
On April 17, 2018, we entered into an amended and restated credit agreement, or the Revolving Credit Agreement, as borrower, with lenders MUFG Union Bank, N.A., as administrative agent, joint lead arranger, joint book runner, swing line lender and letter of credit issuer, and Westpac Banking Corporation, as syndication agent, joint lead arranger and joint book runner. The Revolving Credit Agreement, among other things, provided a senior unsecured revolving credit facility of $800.0 million, with an uncommitted option to increase the revolving credit facility by an additional $300.0 million.
Additionally, on April 17, 2018, ResMed Limited entered into a syndicated facility agreement, or the Term Credit Agreement, as borrower, with lenders MUFG Union Bank, N.A., as administrative agent, joint lead arranger and joint book runner, and Westpac Banking Corporation, as syndication agent, joint lead arranger and joint book runner. The Term Credit Agreement, among other things, provides ResMed Limited a senior unsecured term credit facility of $200.0 million.
On November 5, 2018, we entered into a first amendment to the Revolving Credit Agreement to, among other things, increase the size of our senior unsecured revolving credit facility from $800.0 million to $1.6 billion, with an uncommitted option to increase the revolving credit facility by an additional $300.0 million.
Our obligations under the Revolving Credit Agreement are guaranteed by certain of our direct and indirect U.S. subsidiaries, and ResMed Limited’s obligations under the Term Credit Agreement are guaranteed by us and certain of our direct and indirect U.S. subsidiaries. The Revolving Credit Agreement and Term Credit Agreement contain customary covenants, including, in each case, a financial covenant that requires that we maintain a maximum leverage ratio of funded debt to EBITDA (as defined in the Revolving Credit Agreement and Term Credit Agreement, as applicable). The entire principal amounts of the revolving credit facility and term credit facility, and, in each case, any accrued but unpaid interest may be declared immediately due and payable if an event of default occurs, as defined in the Revolving Credit Agreement and the Term Credit Agreement, as applicable. Events of default under the Revolving Credit Agreement and the Term Credit Agreement include, in each case, failure to make payments when due, the occurrence of a default in the performance of any covenants in the respective agreements or related documents, or certain changes of control of us, or the respective guarantors of the obligations borrowed under the Revolving Credit Agreement and Term Credit Agreement.
The Revolving Credit Agreement and Term Credit Agreement each terminate on April 17, 2023, when all unpaid principal and interest under the loans must be repaid. Amounts borrowed under the Term Credit Agreement will also amortize on a semi-annual basis, with a $6.0 million principal payment required on each such semi-annual amortization date. The outstanding principal amounts will bear interest at a rate equal to LIBOR plus 0.75% to 1.50% (depending on the then-applicable leverage ratio) or the Base Rate (as defined in the Revolving Credit Agreement and the Term Credit Agreement, as applicable) plus 0.0% to 0.50% (depending on the then-applicable leverage ratio). At June 30, 2020, the interest rate that was being charged on the outstanding principal amounts was 1.2%. An applicable commitment fee of 0.100% to 0.175% (depending on the then-applicable leverage ratio) applies on the unused portion of the revolving credit facility. At June 30, 2020, we were in compliance with our debt covenants and there was $680.0 million outstanding under the Revolving Credit Agreement and Term Credit Agreement.
Table of Contents
PART II |
Item 7 |
RESMED INC. AND SUBSIDIARIES
Management’s Discussion and Analysis of Financial Condition and Results of Operations
Senior Notes
On July 10, 2019, we entered into a Note Purchase Agreement with the purchasers to that agreement, in connection with the issuance and sale of $250.0 million principal amount of our 3.24% senior notes due July 10, 2026, and $250.0 million principal amount of our 3.45% senior notes due July 10, 2029. Our obligations under the Note Purchase Agreement and the Notes are unconditionally and irrevocably guaranteed by certain of our direct and indirect U.S. subsidiaries, including ResMed Corp., ResMed Motor Technologies Inc., Birdie Inc., Inova Labs, Inc., Brightree LLC, Brightree Home Health & Hospice LLC, Brightree Patient Collections LLC, ResMed Operations Inc., HEALTHCAREfirst Holding Company, HCF Holdco Company, HEALTHCAREfirst, Inc., CareFacts Information Systems, LLC and Lewis Computer Services, LLC, MatrixCare Holdings Inc., MatrixCare, Inc., Reciprocal Labs Corporation and ResMed SaaS Inc., under a Subsidiary Guaranty Agreement dated as of July 10, 2019. The net proceeds from this transaction were used to pay down borrowings on our Revolving Credit Agreement.
Under the terms of the Note Purchase Agreement, we agreed to customary covenants including with respect to our corporate existence, transactions with affiliates, and mergers and other extraordinary transactions. We also agreed that, subject to limited exceptions, we will maintain a ratio of consolidated funded debt to consolidated EBITDA of no more than 3.50 to 1.00 as of the last day of any fiscal quarter, and will not at any time permit the amount of all secured and unsecured debt of us and our subsidiaries to exceed 10% of our consolidated tangible assets, determined as of the end of our most recently ended fiscal quarter.
On June 30, 2020, we were in compliance with our debt covenants and there was a total of $1,180.0 million outstanding under the Revolving Credit Agreement, Term Credit Agreement and Senior Notes. We expect to satisfy all of our liquidity and long-term debt requirements through a combination of cash on hand, cash generated from operations and undrawn debt facilities.
Critical Accounting Principles and Estimates
The preparation of financial statements in conformity with U.S. GAAP requires us to make estimates and judgments that affect our reported amounts of assets and liabilities, revenues and expenses and related disclosures of contingent assets and liabilities. On an ongoing basis we evaluate our estimates, including those related to allowance for doubtful accounts, inventory reserves, warranty obligations, goodwill, potentially impaired assets, intangible assets, income taxes and contingencies.
We state these accounting policies in the notes to the financial statements and at relevant sections in this discussion and analysis. The estimates are based on the information that is currently available to us and on various other assumptions that we believe to be reasonable under the circumstances. Actual results could vary from those estimates under different assumptions or conditions.
We believe that the following critical accounting policies affect the more significant judgments and estimates used in the preparation of our consolidated financial statements:
(1) Valuation of Goodwill, Intangible and Other Long-Lived Assets. We make assumptions in establishing the carrying value, fair value and estimated lives of our goodwill, intangibles and other long-lived assets. Our goodwill impairment tests are performed at our reporting unit level, which is one level below our operating segments. The criteria used for these evaluations include management’s estimate of the asset’s continuing ability to generate positive income from operations and positive cash flow in future periods compared to the carrying value of the asset, as well as the strategic significance of any identifiable intangible asset in our business objectives. If assets are considered to be impaired, we recognize as impairment the amount by which the carrying value of the assets exceeds their fair value, and for goodwill is limited to the value of goodwill allocated to the impaired reporting unit, as described in Step 1 below. We base useful lives and related amortization or depreciation expense on our estimate of the period that the assets will generate revenues or otherwise be used by us. Factors that would influence the likelihood of a material change in our reported results include significant changes in the asset’s ability to generate positive cash flow, loss of legal ownership or title to the asset, a significant decline in the economic and competitive environment on which the asset depends, significant changes in our strategic business objectives, utilization of the asset, and a significant change in the economic and/or political conditions in certain countries.
We conduct an annual review for goodwill impairment at our reporting unit level based on the following steps:
Step 0 or Qualitative assessment – Evaluate qualitative factors to determine whether it is more likely than not that the fair value of a reporting unit is less than its carrying amount, including goodwill. The factors we consider include, but are not limited to, macroeconomic conditions, industry and market considerations, cost factors, overall financial performance or events-specific to that reporting unit. If or when we determine it is more likely than not that the fair value of a reporting unit is less than the carrying amount, including goodwill, we would move to Step 1 of the quantitative method.
Table of Contents
PART II |
Item 7 |
RESMED INC. AND SUBSIDIARIES
Management’s Discussion and Analysis of Financial Condition and Results of Operations
Step 1 – Compare the fair value for each reporting unit to its carrying value, including goodwill. Fair value is determined based on estimated discounted cash flows. A goodwill impairment charge is recognized for the amount that the carrying amount of a reporting unit, including goodwill, exceeds its fair value, limited to the total amount of goodwill allocated to that reporting unit. If a reporting unit’s fair value exceeds the carrying value, no further work is performed and no impairment charge is necessary.
(2) Income Tax. We assess our income tax positions and record tax benefits for all years subject to audit based upon management’s evaluation of the facts, circumstances and information available at the reporting date. If we determine that it is not more likely than not that we would be able to realize all or part of our net deferred tax assets in the future, an adjustment to the deferred tax assets would be charged to income tax expense in the period such determination is made. Alternatively, if we determine that it is more likely than not that the net deferred tax assets would be realized, any previously provided valuation allowance is reversed. These changes to the valuation allowance and resulting increases or decreases in income tax expense may have a material effect on our operating results.
Our income tax returns are based on calculations and assumptions subject to audit by various tax authorities. In addition, the calculation of our tax liabilities involves dealing with uncertainties in the application of complex tax laws. Although currently immaterial, we recognize liabilities for uncertain tax positions based on a two-step process. The first step is to evaluate the tax position for recognition by determining if the weight of available evidence indicates that it is more likely than not that the position will be sustained on audit, including resolution of related appeals or litigation processes, if any. The second step is to measure the tax benefit as the largest amount that is more than 50% likely of being realized upon settlement. While we believe we have appropriate support for the positions taken on our tax returns, we regularly assess the potential outcomes of examinations by tax authorities in determining the adequacy of our provision for income taxes. Based on our regular assessment, we may adjust the income tax provision and deferred taxes in the period in which the facts that give rise to a revision become known.
In connection with the audit by the ATO for the tax years 2009 to 2013, we received Notices of Amended Assessments in March 2018. Based on these assessments, the ATO asserted that we owe $151.7 million in additional income tax and $38.4 million in accrued interest, of which $75.9 million was paid in April 2018 under a payment arrangement with the ATO. In June 2018, we received a notice from the ATO claiming penalties of 50% of the additional income tax that was assessed or $75.9 million. At June 30, 2020, we recorded a receivable in prepaid taxes and other non-current assets for the amount paid as we ultimately expect this will be refunded by the ATO. The ATO is currently auditing tax years 2014 to 2018. We do not agree with the ATO’s assessments and continue to believe we are more likely than not to be successful in defending our position.
(3) Revenue Recognition. We have determined that we have two operating segments, which are the sleep and respiratory disorders sector of the medical device industry (“Sleep and Respiratory Care”) and the supply of business management software as a service to out-of-hospital health providers (“SaaS”). For products in our Sleep and Respiratory Care business, we transfer control and recognize a sale when products are shipped to the customer in accordance with the contractual shipping terms. For our SaaS business, revenue associated with professional services are recognized as they are provided. We defer the recognition of a portion of the consideration received when performance obligations are not yet satisfied. Consideration received from customers in advance of revenue recognition is classified as deferred revenue. Performance obligations resulting in deferred revenue in our Sleep and Respiratory Care business relate primarily to extended warranties on our devices and the provision of data for patient monitoring. Performance obligations resulting in deferred revenue in our SaaS business relate primarily to the provision of software access with maintenance and support over an agreed term and material rights associated with future discounts upon renewal of some SaaS contracts. Generally, deferred revenue will be recognized over a period of one to five years. Our contracts do not contain significant financing components.
Revenue is measured as the amount of consideration we expect to receive in exchange for transferring goods or providing services. In our Sleep and Respiratory Care business, the amount of consideration received and revenue recognized varies with changes in marketing incentives (e.g., rebates, discounts, free goods) and returns offered to customers. In accounting for these rebate programs, we reduce revenue ratably as sales occur over the rebate period by the expected value of the rebates to be returned to the customer. We also recognize discount on products as a reduction to revenue when control is transferred. We adjust the estimate of revenue for the impact of returned items at the earlier of when the most likely amount of consideration can be estimated, the amount expected to be received changes, or when the consideration becomes fixed. However, returns of products, excluding warranty-related returns, are infrequent and insignificant.
When Sleep and Respiratory Care or SaaS contracts have multiple performance obligations, we generally use an observable price to determine the stand-alone selling price by reference to pricing and discounting practices for the specific product or service when sold separately to similar customers. Revenue is then allocated proportionately, based on the determined stand-alone selling price, to each performance obligation. An allocation is not required for many of our Sleep and Respiratory Care contracts that have a single performance obligation, which is the shipment of our therapy-based equipment.
Table of Contents
PART II |
Item 7 |
RESMED INC. AND SUBSIDIARIES
Management’s Discussion and Analysis of Financial Condition and Results of Operations
Recently Issued Accounting Pronouncements
See Note 3 – New Accounting Pronouncements of the Notes to Consolidated Financial Statements (Part II, Item 8) for a description of recently issued accounting pronouncements, including the expected dates of adoption and estimated effects on our results of operations, financial positions and cash flows.
Off-Balance Sheet Arrangements
As of June 30, 2020, we are not involved in any significant off-balance sheet arrangements, as defined in Item 303(a)(4)(ii) of Regulation S-K promulgated by the SEC.
Table of Contents
PART II |
Item 7A |
RESMED INC. AND SUBSIDIARIES
Quantitative and Qualitative Disclosures About Market and Business Risks
ITEM 7AQUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET AND BUSINESS RISKS
Foreign Currency Market Risk
Our reporting currency is the U.S. dollar, although the financial statements of our non-U.S. subsidiaries are maintained in their respective local currencies. We transact business in various foreign currencies, including a number of major European currencies as well as the Australian dollar. We have significant foreign currency exposure through both our Australian and Singapore manufacturing activities and international sales operations. We have established a foreign currency hedging program using purchased currency options and forward contracts to hedge foreign-currency-denominated financial assets, liabilities and manufacturing cash flows. The goal of this hedging program is to economically manage the financial impact of foreign currency exposures predominantly denominated in euros, Australian dollars and Singapore dollars. Under this program, increases or decreases in our foreign-currency-denominated financial assets, liabilities, and firm commitments are partially offset by gains and losses on the hedging instruments. We do not enter into financial instruments for trading or speculative purposes. The foreign currency derivatives portfolio is recorded in the consolidated balance sheets at fair value and included in Other assets current, Other assets non-current, Accrued expenses and Other liabilities non-current. All movements in the fair value of the foreign currency derivatives are recorded within Other income, net, on our consolidated statements of income.
The table below provides information (in U.S. dollars) on our significant foreign-currency-denominated financial assets by legal entity functional currency as of June 30, 2020 (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
U.S. |
|
|
|
Great Britain |
|
Canadian |
|
Chinese |
|
|
Dollar |
|
Euro |
|
Pound |
|
Dollar |
|
Yuan |
|
|
(USD) |
|
(EUR) |
|
(GBP) |
|
(CAD) |
|
(CNY) |
AUD Functional: |
|
|
|
|
|
|
|
|
|
|
Assets |
|
412,883 |
|
139,175 |
|
- |
|
- |
|
18,949 |
Liability |
|
(414,802) |
|
(120,745) |
|
(309) |
|
- |
|
(564) |
Foreign Currency Hedges |
|
- |
|
(16,852) |
|
- |
|
- |
|
(22,647) |
Net Total |
|
(1,919) |
|
1,578 |
|
(309) |
|
- |
|
(4,262) |
USD Functional: |
|
|
|
|
|
|
|
|
|
|
Assets |
|
- |
|
- |
|
- |
|
19,472 |
|
- |
Liability |
|
- |
|
- |
|
(134) |
|
(5,606) |
|
- |
Foreign Currency Hedges |
|
- |
|
- |
|
- |
|
(14,687) |
|
- |
Net Total |
|
- |
|
- |
|
(134) |
|
(821) |
|
- |
SGD Functional: |
|
|
|
|
|
|
|
|
|
|
Assets |
|
539,940 |
|
145,999 |
|
- |
|
- |
|
12 |
Liability |
|
(238,584) |
|
(67,624) |
|
- |
|
- |
|
- |
Foreign Currency Hedges |
|
(295,000) |
|
(73,025) |
|
- |
|
- |
|
- |
Net Total |
|
6,356 |
|
5,350 |
|
- |
|
- |
|
12 |
Table of Contents
PART II |
Item 7A |
RESMED INC. AND SUBSIDIARIES
Quantitative and Qualitative Disclosures About Market and Business Risks
The table below provides information about our foreign currency derivative financial instruments and presents the information in U.S. dollar equivalents. The table summarizes information on instruments and transactions that are sensitive to foreign currency exchange rates, including foreign currency call options, collars and forward contracts held at June 30, 2020. The table presents the notional amounts and weighted average exchange rates by contractual maturity dates for our foreign currency derivative financial instruments. These notional amounts generally are used to calculate payments to be exchanged under the options contracts (in thousands, except exchange rates):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fair Value Assets / (Liabilities) |
||
Foreign Exchange Contracts |
|
Year 1 |
|
Year 2 |
|
Total |
|
June 30, 2020 |
|
June 30, 2019 |
AUD/USD |
|
|
|
|
|
|
|
|
|
|
Contract amount |
|
- |
|
- |
|
- |
|
- |
|
202 |
Ave. contractual exchange rate |
|
|
|
|
|
|
|
|
|
|
AUD/Euro |
|
|
|
|
|
|
|
|
|
|
Contract amount |
|
61,791 |
|
11,235 |
|
73,026 |
|
886 |
|
(124) |
Ave. contractual exchange rate |
|
AUD 1 = Euro 0.6293 |
|
AUD 1 = Euro 0.58 |
|
AUD 1 = Euro 0.6211 |
|
|
|
|
SGD/Euro |
|
|
|
|
|
|
|
|
|
|
Contract amount |
|
84,260 |
|
5,617 |
|
89,877 |
|
126 |
|
40 |
Ave. contractual exchange rate |
|
SGD 1 = Euro 0.6345 |
|
SGD 1 = Euro 0.6120 |
|
SGD 1 = Euro 0.6331 |
|
|
|
|
SGD/USD |
|
|
|
|
|
|
|
|
|
|
Contract amount |
|
295,000 |
|
- |
|
295,000 |
|
(183) |
|
71 |
Ave. contractual exchange rate |
|
SGD 1 = USD 0.7176 |
|
|
|
SGD 1 = USD 0.7176 |
|
|
|
|
AUD/CNY |
|
|
|
|
|
|
|
|
|
|
Contract amount |
|
22,647 |
|
- |
|
22,647 |
|
(161) |
|
(15) |
Ave. contractual exchange rate |
|
AUD 1 = CNY 4.9450 |
|
|
|
AUD 1 = CNY 4.9450 |
|
|
|
|
USD/CAD |
|
|
|
|
|
|
|
|
|
|
Contract amount |
|
14,687 |
|
- |
|
14,687 |
|
(83) |
|
(66) |
Ave. contractual exchange rate |
|
USD 1 = CAD 1.3695 |
|
|
|
USD 1 = CAD 1.3695 |
|
|
|
|
Interest Rate Risk
We are exposed to risk associated with changes in interest rates affecting the return on our cash and cash equivalents and debt. At June 30, 2020, we held cash and cash equivalents of $463.2 million principally comprising of bank term deposits and at-call accounts and are invested at both short-term fixed interest rates and variable interest rates. At June 30, 2020, there was $680.0 million outstanding under the revolving credit and term loan facilities, which were subject to variable interest rates. A hypothetical 10% change in interest rates during the year ended June 30, 2020, would not have had a material impact on pretax income. We have no interest rate hedging agreements. On July 10, 2019, we entered into the Note Purchase Agreement with the purchasers to that agreement, in connection with the issuance and sale of $250.0 million principal amount of our 3.24% senior notes due July 10, 2026, and $250.0 million principal amount of our 3.45% senior notes due July 10, 2029. The interest rate on these notes is fixed and not subject to fluctuation. Proceeds from the issuance and sale of the notes were used to repay borrowings under the revolving credit facility.
ITEM 8 CONSOLIDATED FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The information required by this Item is incorporated by reference to the financial statements set forth in Item 15 of Part IV of this report, “Exhibits and Consolidated Financial Statement Schedules.”
(a) Index to Consolidated Financial Statements
(b) Supplementary Data
Quarterly Financial Information (unaudited)—The quarterly results for the years ended June 30, 2020 and 2019 are summarized below (in thousands, except per share amounts):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
First |
|
Second |
|
Third |
|
Fourth |
|
Fiscal |
|||||
Net revenue |
|
$ |
681,056 |
|
$ |
736,157 |
|
$ |
769,455 |
|
$ |
770,343 |
|
$ |
2,957,013 |
Gross profit* |
|
|
391,619 |
|
|
427,130 |
|
|
449,662 |
|
|
449,372 |
|
|
1,717,786 |
Net income |
|
|
120,148 |
|
|
160,554 |
|
|
163,137 |
|
|
177,835 |
|
|
621,674 |
Basic earnings per share |
|
|
0.84 |
|
|
1.11 |
|
|
1.13 |
|
|
1.23 |
|
|
4.31 |
Diluted earnings per share |
|
|
0.83 |
|
|
1.10 |
|
|
1.12 |
|
|
1.22 |
|
|
4.27 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2019 |
|
First |
|
Second |
|
Third |
|
Fourth |
|
Fiscal |
|||||
Net revenue |
|
$ |
588,279 |
|
$ |
651,100 |
|
$ |
662,228 |
|
$ |
704,964 |
|
$ |
2,606,572 |
Gross profit* |
|
|
336,138 |
|
|
374,532 |
|
|
380,970 |
|
|
402,432 |
|
|
1,494,071 |
Net income |
|
|
105,737 |
|
|
124,639 |
|
|
105,416 |
|
|
68,797 |
|
|
404,592 |
Basic earnings per share |
|
|
0.74 |
|
|
0.87 |
|
|
0.74 |
|
|
0.48 |
|
|
2.83 |
Diluted earnings per share |
|
|
0.73 |
|
|
0.86 |
|
|
0.73 |
|
|
0.48 |
|
|
2.80 |
*Within our consolidated statements of income for the years ended June 30, 2020 and 2019, cost of sales has been adjusted to include amortization of acquired intangible assets directly applicable to revenue. As a result, gross profit includes amortization of acquired intangible assets relating to cost of sales and operating expenses have been reduced by this amount. There was no impact on income from operations, income before taxes or net income, as a result of this reclassification
Note: the amounts for each quarter are computed independently and, due to the computation formula, the sum of the four quarters may not equal the year.
Report of Independent Registered Public Accounting Firm
To the Stockholders and Board of Directors
ResMed Inc.:
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of ResMed Inc. and subsidiaries (the Company) as of June 30, 2020 and 2019, the related consolidated statements of income, comprehensive income, stockholders’ equity, and cash flows for each of the years in the three year period ended June 30, 2020, and the related notes and financial statement schedule II (collectively, the consolidated financial statements). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of June 30, 2020 and 2019, and the results of its operations and its cash flows for each of the years in the three year period ended June 30, 2020, in conformity with U.S. generally accepted accounting principles.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the Company’s internal control over financial reporting as of June 30, 2020, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission, and our report dated August 12, 2020 expressed an unqualified opinion on the effectiveness of the Company’s internal control over financial reporting.
Change in Accounting Principle
As discussed in Note 3 to the consolidated financial statements, the Company changed its method of accounting for leases beginning July 1, 2019 due to the adoption of the FASB’s Accounting Standards Codification Topic 842, Leases.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matters
The critical audit matters communicated below are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
Evaluation of goodwill triggering events
As discussed in Notes 1(i) and 5 to the consolidated financial statements, the carrying amount of goodwill as of June 30, 2020 was $1,890 million. The Company performs goodwill impairment testing on an annual basis and whenever events or changes in circumstances indicate that the carrying value of a reporting unit, including goodwill, might exceed the fair value of the reporting unit. In the current year, the Company performed qualitative,
or Step 0, assessments to determine whether there was a greater than 50 percent likelihood that the fair value of each reporting unit was less than its carrying value.
We identified the evaluation of goodwill triggering events as a critical audit matter because such events indicate possible impairment of goodwill, which required the application of greater auditor judgment. Potential triggering events, such as macroeconomic conditions, industry and market considerations, cost factors, overall financial performance, market capitalization and events specific to the entity and reporting units, required a higher degree of auditor judgment to evaluate. These possible triggering events could have a significant effect on the Company’s Step 0 assessment and the determination of whether further quantitative analysis of goodwill impairment was required.
The following are the primary procedures we performed to address this critical audit matter. We evaluated the design and tested the operating effectiveness of internal controls related to the critical audit matter. This included a control related to the Company’s assessment of possible goodwill triggering events. We evaluated the Company’s Step 0 assessment for its reporting units by:
Considering macroeconomic indicators such as gross domestic product and inflation by key regions around the world;
Evaluating information from analyst reports in the enterprise software and sleep and respiratory care industries, which are compared to industry and market considerations used by the Company; and
Analyzing information including changes in the costs of raw materials and labor, the financial performance of the reporting units, the Company’s market capitalization, and other entity and reporting-unit specific events.
Evaluation of uncertain tax positions related to Australian Tax Office audits
As discussed in Note 14 to the consolidated financial statements, the Company’s tax filings in Australia for the years 2009 through 2017 are under audit by the Australian Tax Office (ATO). The Company has been assessed $266 million of additional income tax, penalties, and interest for tax years 2009 through 2013 in connection with this tax audit. Certain of these amounts have been paid by the Company to the ATO. However, the Company has not recorded any expense relating to the ongoing audit, or these assessments, as the Company believes it is more likely than not (more than a 50% likelihood) that its tax positions will be upheld.
We identified the evaluation of uncertain tax positions related to Australian Tax Office audits as a critical audit matter. This critical audit matter required challenging auditor judgment due to the nature and the subjectivity of the applicable tax rules and regulations.
The following are the primary procedures we performed to address this critical audit matter. We evaluated the design and tested the operating effectiveness of certain internal controls related to the critical audit matter. This included controls over the Australian tax calculation and assessment of uncertain tax positions. We involved tax professionals with specialized skills and knowledge of Australian tax laws, who assisted in:
Reading formal notices and assessments, and other correspondence received by the Company from the ATO in connection with the audit, as well as responses and information the Company submitted to the ATO in response to its requests for information;
Evaluating the Company’s analysis and conclusions regarding its assertion, which included an assessment of the Company’s analysis of Australian tax laws and regulations related to the specific audit findings, and an evaluation of the facts, assumptions, and representations made; and
Reading legal opinions obtained by the Company from third parties, and inquiring of third-party legal counsel about the likelihood of the Company’s tax position being ultimately upheld.

We have served as the Company’s auditor since 1994.
San Diego, California
August 12, 2020
RESMED INC. AND SUBSIDIARIES
Consolidated Balance Sheets
June 30, 2020 and 2019
(In thousands, except share and per share data)
|
|
|
|
|
|
|
|
|
|
|
|
|
June 30, |
|
June 30, |
||
Assets |
|
|
|
|
|
Current assets: |
|
|
|
|
|
Cash and cash equivalents |
$ |
|
|
$ |
|
Accounts receivable, net of allowance for doubtful accounts of $ |
|
|
|
|
|
Inventories (note 4) |
|
|
|
|
|
Prepaid expenses and other current assets |
|
|
|
|
|
Total current assets |
|
|
|
|
|
Non-current assets: |
|
|
|
|
|
Property, plant and equipment, net (note 4) |
|
|
|
|
|
Operating lease right-of-use assets (note 10) |
|
|
|
|
- |
Goodwill (note 5) |
|
|
|
|
|
Other intangible assets, net (note 5) |
|
|
|
|
|
Deferred income taxes (note 14) |
|
|
|
|
|
Prepaid taxes and other non-current assets |
|
|
|
|
|
Total non-current assets |
|
|
|
|
|
Total assets |
$ |
|
|
$ |
|
Liabilities and Stockholders’ Equity |
|
|
|
|
|
Current liabilities: |
|
|
|
|
|
Accounts payable |
$ |
|
|
$ |
|
Accrued expenses (note 7) |
|
|
|
|
|
Operating lease liabilities, current (note 10) |
|
|
|
|
- |
Deferred revenue |
|
|
|
|
|
Income taxes payable (note 14) |
|
|
|
|
|
Short-term debt, net (note 9) |
|
|
|
|
|
Total current liabilities |
|
|
|
|
|
Non-current liabilities: |
|
|
|
|
|
Deferred revenue |
|
|
|
|
|
Deferred income taxes (note 14) |
|
|
|
|
|
Operating lease liabilities, non-current (note 10) |
|
|
|
|
- |
Other long-term liabilities |
|
|
|
|
|
Long-term debt, net (note 9) |
|
|
|
|
|
Long-term income taxes payable (note 14) |
|
|
|
|
|
Total non-current liabilities |
|
|
|
|
|
Total liabilities |
|
|
|
|
|
Commitments and contingencies (note 17) |
|
|
|
||
Stockholders’ equity: (note 11) |
|
|
|
|
|
Preferred stock, $ |
|
|
|
|
|
Common stock, $ |
|
|
|
|
|
Additional paid-in capital |
|
|
|
|
|
Retained earnings |
|
|
|
|
|
Treasury stock, at cost, |
|
( |
|
|
( |
Accumulated other comprehensive loss |
|
( |
|
|
( |
Total stockholders’ equity |
|
|
|
|
|
Total liabilities and stockholders’ equity |
$ |
|
|
$ |
|
See accompanying notes to consolidated financial statements.
RESMED INC. AND SUBSIDIARIES
Consolidated Statements of Income
Years Ended June 30, 2020, 2019 and 2018
(In thousands, except per share data)
|
|
|
|
|
|
|
|
|
|
|
|
June 30, 2020 |
|
June 30, 2019 |
|
June 30, 2018 |
|||
Net revenue - Sleep and Respiratory Care products |
|
$ |
|
|
$ |
|
|
$ |
|
Net revenue - Software as a Service |
|
|
|
|
|
|
|
|
|
Net revenue |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cost of sales - Sleep and Respiratory Care products |
|
|
|
|
|
|
|
|
|
Cost of sales - Software as a Service |
|
|
|
|
|
|
|
|
|
Cost of sales |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Amortization of acquired intangible assets - Sleep and Respiratory Care products |
|
|
|
|
|
|
|
|
|
Amortization of acquired intangible assets - Software as a Service |
|
|
|
|
|
|
|
|
|
Amortization of acquired intangible assets |
|
|
|
|
|
|
|
|
|
Total cost of sales |
|
|
|
|
|
|
|
|
|
Gross profit |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Selling, general and administrative |
|
|
|
|
|
|
|
|
|
Research and development |
|
|
|
|
|
|
|
|
|
Amortization of acquired intangible assets |
|
|
|
|
|
|
|
|
|
Restructuring expenses (note 19) |
|
|
- |
|
|
|
|
|
|
Litigation settlement expenses (note 20) |
|
|
( |
|
|
|
|
|
- |
Acquisition related expenses (note 18) |
|
|
- |
|
|
|
|
|
- |
Total operating expenses |
|
|
|
|
|
|
|
|
|
Income from operations |
|
|
|
|
|
|
|
|
|
Other income (loss), net: |
|
|
|
|
|
|
|
|
|
Interest income |
|
|
|
|
|
|
|
|
|
Interest expense |
|
|
( |
|
|
( |
|
|
( |
Loss attributable to equity method investments (note 6) |
|
|
( |
|
|
( |
|
|
- |
Other, net (note 13) |
|
|
( |
|
|
( |
|
|
( |
Total other income (loss), net |
|
|
( |
|
|
( |
|
|
( |
Income before income taxes |
|
|
|
|
|
|
|
|
|
Income taxes |
|
|
|
|
|
|
|
|
|
Net income |
|
$ |
|
|
$ |
|
|
$ |
|
|
|
|
|
|
|
|
|
|
|
Basic earnings per share (note 12) |
|
$ |
|
|
$ |
|
|
$ |
|
Diluted earnings per share (note 12) |
|
$ |
|
|
$ |
|
|
$ |
|
Dividend declared per share |
|
$ |
|
|
$ |
|
|
$ |
|
Basic shares outstanding (000's) |
|
|
|
|
|
|
|
|
|
Diluted shares outstanding (000's) |
|
|
|
|
|
|
|
|
|
See accompanying notes to consolidated financial statements.
RESMED INC. AND SUBSIDIARIES
Consolidated Statements of Comprehensive Income
Years Ended June 30, 2020, 2019 and 2018
(In US$ thousands)
|
|
|
|
|
|
|
|
|
|
|
|
Years Ended June 30, |
|||||||
|
|
2020 |
|
2019 |
|
2018 |
|||
Net income |
|
$ |
|
|
|
|
|
|
|
Other comprehensive (loss) income: |
|
|
|
|
|
|
|
|
|
Foreign currency translation (loss) gain adjustments |
|
|
( |
|
|
( |
|
|
( |
Comprehensive income |
|
$ |
|
|
$ |
|
|
$ |
|
See accompanying notes to consolidated financial statements.
RESMED INC. AND SUBSIDIARIES
Consolidated Statements of Stockholders’ Equity
Years ended June 30, 2020, 2019 and 2018
(In thousands)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Common Stock |
Additional |
Treasury Stock |
Retained |
Accumulated |
|
|
|||||||
|
Shares |
Amount |
Capital |
Shares |
Amount |
Earnings |
Income (Loss) |
Total |
||||||
Balance, June 30, 2017 |
|
$ |
|
$ |
|
( |
$ |
( |
$ |
|
$ |
( |
$ |
|
Common stock issued on exercise of options (note 11) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Common stock issued on vesting of restricted stock units, net of shares withheld for tax (note 11) |
|
|
|
|
( |
|
|
|
|
|
|
|
|
( |
Common stock issued on employee stock purchase plan (note 11) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Treasury stock purchases |
|
|
( |
|
|
( |
|
( |
|
|
|
|
|
( |
Stock-based compensation costs |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other comprehensive income (loss) |
|
|
|
|
|
|
|
|
|
|
|
( |
|
( |
Net income |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dividends declared |
|
|
|
|
|
|
|
|
|
( |
|
|
|
( |
Balance, June 30, 2018 |
|
$ |
|
$ |
|
( |
$ |
( |
$ |
|
$ |
( |
$ |
|
Common stock issued on exercise of options (note 11) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Common stock issued on vesting of restricted stock units, net of shares withheld for tax (note 11) |
|
|
|
|
( |
|
|
|
|
|
|
|
|
( |
Common stock issued on employee stock purchase plan (note 11) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Treasury stock purchases |
|
|
( |
|
|
( |
|
( |
|
|
|
|
|
( |
Stock-based compensation costs |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other comprehensive income (loss) |
|
|
|
|
|
|
|
|
|
|
|
( |
|
( |
Net income |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Cumulative effect of change in accounting standards |
|
|
|
|
|
|
|
|
|
( |
|
|
|
( |
Dividends declared |
|
|
|
|
|
|
|
|
|
( |
|
|
|
( |
Balance, June 30, 2019 |
|
$ |
|
$ |
|
( |
$ |
( |
$ |
|
$ |
( |
$ |
|
Common stock issued on exercise of options (note 11) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Common stock issued on vesting of restricted stock units, net of shares withheld for tax (note 11) |
|
|
|
|
( |
|
|
|
|
|
|
|
|
( |
Common stock issued on employee stock purchase plan (note 11) |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Treasury stock purchases |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stock-based compensation costs |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Other comprehensive income (loss) |
|
|
|
|
|
|
|
|
|
|
|
( |
|
( |
Net income |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dividends declared |
|
|
|
|
|
|
|
|
|
( |
|
|
|
( |
Balance, June 30, 2020 |
|
$ |
|
$ |
|
( |
$ |
( |
$ |
|
$ |
( |
$ |
|
See accompanying notes to consolidated financial statements.
RESMED INC. AND SUBSIDIARIES
Consolidated Statements of Cash Flows
Years ended June 30, 2020, 2019 and 2018
(In thousands)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
June 30, 2020 |
|
June 30, 2019 |
|
June 30, 2018 |
|||
Cash flows from operating activities: |
|
|
|
|
|
|
|
|
|
Net income |
|
$ |
|
|
$ |
|
|
$ |
|
Adjustment to reconcile net income to net cash provided by operating activities: |
|
|
|
|
|
|
|
|
|
Depreciation and amortization |
|
|
|
|
|
|
|
|
|
Amortization of right-of-use-assets |
|
|
|
|
|
- |
|
|
- |
Stock-based compensation costs |
|
|
|
|
|
|
|
|
|
Loss attributable to equity method investments (note 6) |
|
|
|
|
|
|
|
|
- |
Impairment of equity investments (note 6) |
|
|
|
|
|
|
|
|
|
Gain on previously held equity interest (note 18) |
|
|
- |
|
|
( |
|
|
- |
Changes in fair value of business combination contingent consideration |
|
|
( |
|
|
( |
|
|
|
Changes in operating assets and liabilities, net of effect of acquisitions: |
|
|
|
|
|
|
|
|
|
Accounts receivable |
|
|
|
|
|
( |
|
|
( |
Inventories |
|
|
( |
|
|
( |
|
|
|
Prepaid expenses, net deferred income taxes and other current assets |
|
|
( |
|
|
( |
|
|
( |
Accounts payable, accrued expenses and other |
|
|
( |
|
|
( |
|
|
|
Net cash provided by operating activities |
|
|
|
|
|
|
|
|
|
Cash flows from investing activities: |
|
|
|
|
|
|
|
|
|
Purchases of property, plant and equipment |
|
|
( |
|
|
( |
|
|
( |
Patent registration costs |
|
|
( |
|
|
( |
|
|
( |
Business acquisitions, net of cash acquired |
|
|
( |
|
|
( |
|
|
( |
Purchases of investments (note 6) |
|
|
( |
|
|
( |
|
|
( |
Payments on maturity of foreign currency contracts |
|
|
( |
|
|
( |
|
|
( |
Net cash used in investing activities |
|
|
( |
|
|
( |
|
|
( |
Cash flows from financing activities: |
|
|
|
|
|
|
|
|
|
Proceeds from issuance of common stock, net |
|
|
|
|
|
|
|
|
|
Taxes paid related to net share settlement of equity awards |
|
|
( |
|
|
( |
|
|
( |
Purchases of treasury stock |
|
|
- |
|
|
( |
|
|
( |
Payments of business combination contingent consideration |
|
|
( |
|
|
( |
|
|
( |
Proceeds from borrowings, net of borrowing costs |
|
|
|
|
|
|
|
|
|
Repayment of borrowings |
|
|
( |
|
|
( |
|
|
( |
Dividends paid |
|
|
( |
|
|
( |
|
|
( |
Net cash provided by (used in) financing activities |
|
|
( |
|
|
|
|
|
( |
Effect of exchange rate changes on cash |
|
|
|
|
|
( |
|
|
( |
Net increase (decrease) in cash and cash equivalents |
|
|
|
|
|
( |
|
|
( |
Cash and cash equivalents at beginning of period |
|
|
|
|
|
|
|
|
|
Cash and cash equivalents at end of period |
|
$ |
|
|
$ |
|
|
$ |
|
Supplemental disclosure of cash flow information: |
|
|
|
|
|
|
|
|
|
Income taxes paid, net of refunds |
|
$ |
|
|
$ |
|
|
$ |
|
Interest paid |
|
$ |
|
|
$ |
|
|
$ |
|
Fair value of assets acquired, excluding cash |
|
$ |
|
|
$ |
|
|
$ |
|
Liabilities assumed |
|
|
( |
|
|
( |
|
|
- |
Goodwill on acquisition |
|
|
|
|
|
|
|
|
|
Deferred payments |
|
|
|
|
|
( |
|
|
|
Fair value of contingent consideration |
|
|
( |
|
|
- |
|
|
- |
Cash paid for acquisitions |
|
$ |
|
|
$ |
|
|
$ |
|
See accompanying notes to consolidated financial statements.
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
ResMed Inc. (referred to herein as “we”, “us”, “our” or the “Company”) is a Delaware corporation formed in March 1994 as a holding company for the ResMed Group. Through our subsidiaries, we design, manufacture and market equipment for the diagnosis and treatment of sleep-disordered breathing and other respiratory disorders, including obstructive sleep apnea. Our manufacturing operations are located in Australia, Singapore, Malaysia, France, China and the United States. Major distribution and sales sites are located in the United States, Germany, France, the United Kingdom, Switzerland, Australia, Japan, China, Finland, Norway and Sweden. We also operate a Software as a Service (“SaaS”) business in the United States that includes out-of-hospital software platforms designed to support the professionals and caregivers who help people stay healthy in the home or care setting of their choice.
(a)
The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries. All significant inter-company transactions and balances have been eliminated in consolidation. The preparation of financial statements in conformity with U.S. generally accounting principles requires management estimates and assumptions that affect amounts reported in the consolidated financial statements and accompanying notes. Actual results could differ from management’s estimates.
(b)
In accordance with Accounting Standard Codification (“ASC”) Topic 606, “Revenue from Contracts with Customers”, we account for a contract with a customer when there is a legally enforceable contract, the rights of the parties are identified, the contract has commercial substance, and collectability of the contract consideration is probable. We have determined that we have two operating segments, which are the sleep and respiratory disorders sector of the medical device industry (“Sleep and Respiratory Care”) and the supply of business management software as a service to out-of-hospital health providers (“SaaS”). Our Sleep and Respiratory Care revenue relates primarily to the sale of our products that are therapy-based equipment. Some contracts include additional performance obligations such as the provision of extended warranties and data for patient monitoring. Our SaaS revenue relates to the provision of software access with ongoing support and maintenance services as well as professional services such as training and consulting.
Disaggregation of revenue
See note 15 – Segment Information for our net revenue disaggregated by segment, product and region for the years ended June 30, 2020, 2019 and 2018.
Effective in the fourth quarter of the fiscal year ended June 30, 2020, our consolidated statements of income separately present the revenues and related costs of the Sleep and Respiratory Care and SaaS segments. Net revenues and cost of sales were previously presented on an aggregate basis. This change separately states net sales of products and revenues from services, which materially aligns with net revenues associated with our Sleep and Respiratory Care and SaaS segments, respectively. While this change has been applied retrospectively to the consolidated statements of income for the years ended June 30, 2019 and 2018, there was no impact on net revenue, cost of sales, income from operations, income before taxes or net income as a result of this change.
Performance obligations and contract balances
Revenue is recognized when performance obligations under the terms of a contract with a customer are satisfied; generally, this occurs with the transfer of risk and/or control of our products are provided at a point in time. For products in our Sleep and Respiratory Care business, we transfer control and recognize a sale when products are shipped to the customer in accordance with the contractual shipping terms. For our SaaS business, revenue associated with professional services are recognized as they are provided. We defer the recognition of a portion of the consideration received when performance obligations are not yet satisfied. Consideration received from customers in advance of revenue recognition is classified as deferred revenue. Performance obligations resulting in deferred revenue in our Sleep and Respiratory Care business relate primarily to extended warranties on our devices and the provision of data for patient monitoring. Performance obligations resulting in deferred revenue in our SaaS business relate primarily to the provision of software access with maintenance and support over an agreed term and material rights associated with future discounts upon renewal of some SaaS contracts. Generally, deferred revenue will be recognized over a period of
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
The following table summarizes our contract balances as of June 30, 2020 and 2019 (in thousands):
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
|
Balance sheet caption |
||
Contract assets |
|
|
|
|
|
|
|
|
Accounts receivable, net |
|
$ |
|
|
$ |
|
|
Accounts receivable, net |
Unbilled revenue, current |
|
|
|
|
|
|
|
Prepaid expenses and other current assets |
Unbilled revenue, non-current |
|
|
|
|
|
|
|
Prepaid taxes and other non-current assets |
|
|
|
|
|
|
|
|
|
Contract liabilities |
|
|
|
|
|
|
|
|
Deferred revenue, current |
|
|
( |
|
|
( |
|
Deferred revenue (current liabilities) |
Deferred revenue, non-current |
|
|
( |
|
|
( |
|
Deferred revenue (non-current liabilities) |
Transaction price determination
Revenue is measured as the amount of consideration we expect to receive in exchange for transferring goods or providing services. In our Sleep and Respiratory Care segment, the amount of consideration received and revenue recognized varies with changes in marketing incentives (e.g., rebates, discounts, free goods) and returns offered to customers and their customers. When we give customers the right to return eligible products and receive credit, returns are estimated based on an analysis of historical experience. However, returns of products, excluding warranty-related returns, are infrequent and insignificant. We adjust the estimate of revenue at the earlier of when the most likely amount of consideration can be estimated, the amount expected to be received changes, or when the consideration becomes fixed.
We offer our Sleep and Respiratory Care customers cash or product rebates based on volume or sales targets measured over quarterly or annual periods. We estimate rebates based on each customer’s expected achievement of its targets. In accounting for these rebate programs, we reduce revenue ratably as sales occur over the rebate period by the expected value of the rebates to be returned to the customer. Rebates measured over a quarterly period are updated based on actual sales results and, therefore, no estimation is required to determine the reduction to revenue. For rebates measured over annual periods, we update our estimates on a quarterly basis based on actual sales results and updated forecasts for the remaining rebate periods. We also offer discounts to both our Sleep and Respiratory Care as well as our SaaS customers as part of normal business practice and these are deducted from revenue when the sale occurs.
When Sleep and Respiratory Care or SaaS contracts have multiple performance obligations, we generally use an observable price to determine the stand-alone selling price by reference to pricing and discounting practices for the specific product or service when sold separately to similar customers. Revenue is then allocated proportionately, based on the determined stand-alone selling price, to each performance obligation. An allocation is not required for many of our Sleep and Respiratory Care contracts that have a single performance obligation, which is the shipment of our therapy-based equipment.
Accounting and practical expedient elections
We have elected to account for shipping and handling activities associated with our Sleep and Respiratory Care segment as a fulfillment cost within cost of sales, and record shipping and handling costs collected from customers in net revenue. We have also elected for all taxes assessed by government authorities that are imposed on and concurrent with revenue-producing transactions, such as sales and value added taxes, to be excluded from revenue and presented on a net basis. We have adopted two practical expedients including the “right to invoice” practical expedient, which allows us to recognize revenue in the amount of the invoice when it corresponds directly with the value of performance completed to date and which is relevant for some of our SaaS contracts. The second practical expedient adopted permits relief from considering a significant financing component when the payment for the good or service is expected to be one year or less.
(c)
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(d)
The fair value of financial instruments is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. We measure our financial instruments at fair value at each reporting period using a fair value hierarchy that requires that we maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value. A financial instrument’s classification within the fair value hierarchy is based upon the lowest level of input that is significant to the fair value measurement. Three levels of inputs may be used to measure fair value:
Level 1 - Observable inputs that reflect quoted prices (unadjusted) for identical assets or liabilities in active markets.
Level 2 - Other inputs that are directly or indirectly observable in the marketplace.
Level 3 - Unobservable inputs that are supported by little or no market activity.
(e)
Cash equivalents include certificates of deposit and other highly liquid investments and we state them at cost, which approximates market. We consider investments with original maturities of 90 days or less to be cash equivalents for purposes of the consolidated statements of cash flows.
(f)
We state inventories at the lower of cost (determined principally by the first-in, first-out method) or net realizable value. We include material, labor and manufacturing overhead costs in finished goods and work-in-process inventories. We review and provide for any product obsolescence in our manufacturing and distribution operations by assessing throughout the year individual products and components (based on estimated future usage and sales).
(g)
We record property, plant and equipment, including rental and demonstration equipment at cost. We compute depreciation expense using the straight-line method over the estimated useful lives of the assets. Useful lives are generally
Depreciation expense for property, plant, and equipment was $
(h)
We capitalize the registration costs for new patents and amortize the costs over the estimated useful life of the patent, which is generally ten years. If a patent is superseded or a product is retired, any unamortized costs are written off immediately.
We amortize all of our other intangible assets on a straight-line basis over their estimated useful lives, which range from
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(i)
We conducted our annual review for goodwill impairment during the final quarter of 2020. Our goodwill impairment review is performed at our reporting unit level, which is one level below our operating segments and involves the following steps:
Step 0 or Qualitative assessment – Evaluate qualitative factors to determine whether it is more likely than not that the fair value of a reporting unit is less than its carrying amount, including goodwill. The factors we consider include, but are not limited to, macroeconomic conditions, industry and market considerations, cost factors, overall financial performance or events-specific to that reporting unit. If or when we determine it is more likely than not that the fair value of a reporting unit is less than the carrying amount, including goodwill, we would move to Step 1 of the quantitative method.
Step 1 – Compare the fair value for each reporting unit to its carrying value, including goodwill. Fair value is determined based on estimated discounted cash flows. A goodwill impairment charge is recognized for the amount that the carrying amount of a reporting unit, including goodwill, exceeds its fair value, limited to the total amount of goodwill allocated to that reporting unit. If a reporting unit’s fair value exceeds the carrying value, no further work is performed and no impairment charge is necessary.
(j)
Equity investments whereby we have significant influence but not control over the investee, and are not the primary beneficiary of the investee’s activities, are accounted for under the equity method. Under this method, we record our share of gains or losses attributable to equity method investments.
Non-marketable equity securities consist of investments in privately held companies without readily determinable fair values, and are reported at cost, minus impairment, if any, plus or minus changes resulting from observable price changes in orderly transactions for the identical or similar investment of the same issuer. We estimate the fair value of our equity investments using Level 3 inputs to assess whether impairment losses shall be recorded.
(k)
We record all research and development expenses in the period we incur them.
(l)
The consolidated financial statements of our non-U.S. subsidiaries, whose functional currencies are other than the U.S. dollar, are translated into U.S. dollars for financial reporting purposes. We translate assets and liabilities of non-U.S. subsidiaries whose functional currencies are other than the U.S. dollar at period end exchange rates, but translate revenue and expense transactions at average exchange rates for the period. We recognize cumulative translation adjustments as part of comprehensive income, as detailed in the consolidated statements of comprehensive income, and include those adjustments in accumulated other comprehensive income in the consolidated balance sheets until such time the relevant subsidiary is sold or substantially or completely liquidated. We reflect gains and losses on transactions denominated in other than the functional currency of an entity in our results of operations.
(m)
We transact business in various foreign currencies, including a number of major European currencies as well as the Australian and Singapore dollars. We have significant foreign currency exposure through both our Australian and Singaporean manufacturing activities, and international sales operations. We have established a foreign currency hedging program using purchased currency options and forward contracts to hedge foreign-currency-denominated financial assets, liabilities and manufacturing cash flows. The terms of such foreign currency hedging contracts generally do not exceed three years. The goal of this hedging program is to economically manage the financial impact of foreign currency exposures denominated mainly in Euros, Australian and Singapore dollars. Under this program, increases or decreases in our foreign currency denominated financial assets, liabilities, and firm commitments are partially offset by gains and losses on the hedging instruments.
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
We do not designate these foreign currency contracts as hedges. We have determined our hedge program to be a non-effective hedge as defined under the FASB issued authoritative guidance. All movements in the fair value of the foreign currency instruments are recorded within other income, net in our consolidated statements of income and through changes in our operating assets and liabilities within our consolidated statements of cash flows. We classify purchases of foreign currency derivatives and proceeds received from the exercise of foreign currency derivatives as an investing activity within our consolidated statements of cash flows. We do not enter into financial instruments for trading or speculative purposes.
We held foreign currency instruments with notional amounts totaling $
(n)
We account for income taxes under the asset and liability method. We recognize deferred tax assets and liabilities for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. We measure deferred tax assets and liabilities using the enacted tax rates we expect to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date.
We recognize the impact of a tax position in the consolidated financial statements only if that position is more likely than not of being sustained upon examination by taxing authorities, based on the technical merits of the position. Any interest and penalties related to uncertain tax positions are reflected in income tax expense.
(o)
We provide for the estimated cost of product warranties on our Sleep and Respiratory Care products at the time the related revenue is recognized. We determine the amount of this provision by using a financial model, which takes into consideration actual historical expenses and potential risks associated with our different products. We use this financial model to calculate the future probable expenses related to warranty and the required level of the warranty provision. Although we engage in product improvement programs and processes, our warranty obligation is affected by product failure rates and costs incurred to correct those product failures. Should actual product failure rates or estimated costs to repair those product failures differ from our estimates, we would be required to revise our estimated warranty provision.
(p)
We maintain an allowance for doubtful accounts for estimated losses resulting from the inability of our customers to make required payments, which results in bad debt expense. We determine the adequacy of this allowance by periodically evaluating individual customer receivables, considering a customer’s financial condition, credit history and current economic conditions. Customer receivables are charged against the allowance when they are deemed uncollectible. We are also contingently liable, within certain limits, in the event of a customer default, to independent financing companies in connection with customer financing programs. We monitor the collection status of these installment receivables and provide for estimated losses separately under accrued expenses within our consolidated balance sheets based upon our historical collection experience with such receivables and a current assessment of our credit exposure.
(q)
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
(r)
We record a liability in the consolidated financial statements for loss contingencies when a loss is known or considered probable and the amount can be reasonably estimated. If the reasonable estimate of a known or probable loss is a range, and no amount within the range is a better estimate than any other, the minimum amount of the range is accrued. If a loss is reasonably possible but not known or probable, and can be reasonably estimated, the estimated loss or range of loss is disclosed. When determining the estimated loss or range of loss, significant judgment is required to estimate the amount and timing of a loss to be recorded.
(a) Recently issued accounting standards not yet adopted
ASU No. 2016-13 “Financial Instruments - Credit Losses: Measurement of Credit Losses on Financial Instruments”
In June 2016, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2016-13, “Financial Instruments - Credit Losses: Measurement of Credit Losses on Financial Instruments” (Topic 326), which amends the impairment model by requiring entities to use a forward-looking approach based on expected losses to estimate credit losses on certain types of financial instruments, including trade receivables. The guidance is effective for us beginning in the first quarter of the fiscal year ending June 30, 2021 and will be adopted using a modified retrospective approach, with a cumulative-effect adjustment recorded directly to retained earnings. We do not expect the adoption to have a material impact on our consolidated financial statements.
ASU No. 2018-15 “Intangibles-Goodwill and Other-Internal-Use Software: Customer’s Accounting for Implementation Costs Incurred in a Cloud Computing Arrangement That Is a Service Contract”
In August 2018, the FASB issued ASU No. 2018-15, “Intangibles-Goodwill and Other-Internal-Use Software: Customer’s Accounting for Implementation Costs Incurred in a Cloud Computing Arrangement That Is a Service Contract” (Subtopic 350-40), which aligns the requirements for capitalizing implementation costs incurred in a hosting arrangement that is a service contract with the requirements for capitalizing implementation costs incurred to develop or obtain internal-use software. The guidance is effective for us beginning in the first quarter of the fiscal year ending June 30, 2021 and will be applied prospectively. Under the new ASU, capitalized implementation costs will be presented as other non-current assets on our consolidated balance sheets and within operating cash flows on our consolidated statements of cash flows. The adoption of this ASU is not expected to have a material effect on our consolidated financial statements.
ASU No. 2020-04 “Reference Rate Reform: Facilitation of the Effects of Reference Rate Reform on Financial Reporting”
In March 2020, the FASB issued ASU No. 2020-04, “Facilitation of the Effects of Reference Rate Reform on Financial Reporting” (Topic 848), which provides optional expedients and exceptions for applying U.S. GAAP to contracts, hedging relationships, and other transactions that reference LIBOR or another reference rate expected to be discontinued because of reference rate reform. The guidance is effective for us as of March 12, 2020 through December 31, 2022. We are currently evaluating the impact that this guidance, if elected, will have on our consolidated financial statements.
(b) Recently adopted accounting pronouncements
ASU No. 2016-02, “Leases”
In February 2016, the FASB issued ASU No. 2016-02, “Leases” (Topic 842). Under the new guidance, lessees are required to recognize a right-of-use asset (“ROU”) and a lease liability on the balance sheet for all leases, other than those that meet the definition of a short-term lease. This update establishes a lease asset and lease liability by lessees for those leases classified as operating under prior GAAP. Leases are classified as either operating or finance under the new guidance. Operating leases result in straight-line expense in the income statement, similar to prior operating lease treatment, and finance leases result in more expense being recognized in the earlier years of the lease term, similar to prior capital lease treatment. For lessors, the update more closely aligns lease accounting to comparable guidance in the new revenue standards described.
Effective, July 1, 2019, we adopted the new standard on a modified retrospective transition basis for leases existing at, or entered into after, the date of adoption. In addition, we elected the package of practical expedients permitted under the transition guidance to not reassess (1) whether any expired or existing contracts are, or contain, leases, (2) the lease classification for expired or existing leases, and (3) initial direct costs for existing leases. In preparation for and upon adoption of this guidance, we have designed and operated internal controls over its implementation, which includes a system solution for lease administration, accounting and disclosures of financial information surrounding our leasing arrangements.
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
The adoption of the guidance on July 1, 2019 resulted in the recognition of ROU assets of $
ASU No. 2017-04 “Intangibles-Goodwill and Other: Simplifying the Test for Goodwill Impairment”
In January 2017, the FASB issued ASU No. 2017-04, “Intangibles-Goodwill and Other: Simplifying the Test for Goodwill Impairment” (Topic 350). ASU 2017-04 eliminates step two of the goodwill impairment test and specifies that goodwill impairment should be measured by comparing the fair value of a reporting unit with its carrying amount. Additionally, the amount of goodwill allocated to each reporting unit with a zero or negative carrying amount of net assets should be disclosed. We adopted this guidance in the fourth quarter of fiscal year June 30, 2020. The adoption did not have a material impact on our consolidated financial statements.
(c) Adjustment to prior periods
As noted at note 2b) – Disaggregation of revenue, we now present revenue and cost of sales for our Sleep and Respiratory Care and SaaS segments on the consolidated statements of income. Additionally, within our consolidated statements of income for the years ended June 30, 2020, 2019 and 2018, cost of sales has been adjusted to include amortization of acquired intangible assets directly applicable to revenue. As a result, gross profit now includes amortization of acquired intangible assets relating to cost of sales and operating expenses have been reduced by this amount. There was no impact on income from operations, income before taxes or net income, as a result of this reclassification. The adjustments to the previously reported amounts are not material.
The table below presents a reconciliation of amortization of acquired intangible assets by income statement caption summing to total amortization of acquired intangible assets as previously reported for the years ended June 30, 2019 and June 30, 2018 (in thousands):
|
|
|
|
|
|
|
|
|
2019 |
|
2018 |
||
Amortization of acquired intangible assets related to cost of sales |
|
$ |
|
|
$ |
|
Amortization of acquired intangible assets related to operating expenses |
|
|
|
|
|
|
Total as previously reported |
|
$ |
|
|
$ |
|
The table below presents a reconciliation of gross profit as previously reported for the years ended June 30, 2019 and June 30, 2018 adjusted for the amortization of acquired intangible assets now included in cost of sales (in thousands):
|
|
|
|
|
|
|
|
|
2019 |
|
2018 |
||
Gross profit as previously reported |
|
$ |
|
|
$ |
|
Amortization of acquired intangible assets related to cost of sales |
|
|
( |
|
|
( |
Gross profit |
|
$ |
|
|
$ |
|
Components of selected captions in the consolidated balance sheets consisted of the following as of June 30, 2020 and June 30, 2019 (in thousands):
|
|
|
|
|
|
|
Inventories |
|
2020 |
|
2019 |
||
Raw materials |
|
$ |
|
|
$ |
|
Work in progress |
|
|
|
|
|
|
Finished goods |
|
|
|
|
|
|
Total inventories |
|
$ |
|
|
$ |
|
|
|
|
|
|
|
|
Property, Plant and Equipment |
|
2020 |
|
2019 |
||
Machinery and equipment |
|
$ |
|
|
$ |
|
Computer equipment |
|
|
|
|
|
|
Furniture and fixtures |
|
|
|
|
|
|
Vehicles |
|
|
|
|
|
|
Clinical, demonstration and rental equipment |
|
|
|
|
|
|
Leasehold improvements |
|
|
|
|
|
|
Land |
|
|
|
|
|
|
Buildings |
|
|
|
|
|
|
Property, plant and equipment, at cost |
|
$ |
|
|
$ |
|
Accumulated depreciation and amortization |
|
|
( |
|
|
( |
Property, plant and equipment, net |
|
$ |
|
|
$ |
|
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
Goodwill
For each of the years ended June 30, 2020 and June 30, 2019, we have
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|||||||
|
|
Sleep and |
|
SaaS |
|
Total |
|||
Balance at the beginning of the period |
|
$ |
|
|
$ |
|
|
$ |
|
Business acquisitions |
|
|
|
|
|
|
|
|
|
Adjustment to fair values of preliminary purchase price allocations |
|
|
|
|
|
|
|
|
|
Foreign currency translation adjustments |
|
|
( |
|
|
|
|
|
( |
Balance at the end of the period |
|
$ |
|
|
$ |
|
|
$ |
|
Other Intangible Assets
Other intangibles, net are comprised of the following as of June 30, 2020 and June 30, 2019 (in thousands):
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
||
Developed/core product technology |
|
$ |
|
|
$ |
|
Accumulated amortization |
|
|
( |
|
|
( |
Developed/core product technology, net |
|
|
|
|
|
|
Customer relationships |
|
|
|
|
|
|
Accumulated amortization |
|
|
( |
|
|
( |
Customer relationships, net |
|
|
|
|
|
|
Other intangibles |
|
|
|
|
|
|
Accumulated amortization |
|
|
( |
|
|
( |
Other intangibles, net |
|
|
|
|
|
|
Total other intangibles, net |
|
$ |
|
|
$ |
|
Intangible assets consist of developed/core product technology, trade names, non-compete agreements, customer relationships, and patents, and we amortize them over the estimated useful life of the assets, generally between
Refer to note 18 of the consolidated financial statements for details of acquisitions.
Amortization expense related to identified intangible assets for the years ended June 30, 2020 and June 30, 2019 was $
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fiscal Years Ending June 30 |
|||||||||||||
|
|
2021 |
|
2022 |
|
2023 |
|
2024 |
|
2025 |
|||||
Estimated amortization expense |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
We have a number of equity investments in privately held companies that are unconsolidated entities and are recorded in the non-current balance of other assets on the consolidated balance sheets. The following table shows a reconciliation of the changes in all of our investments during the years ended June 30, 2020 and June 30, 2019 (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
||
Equity method investments |
|
|
|
|
|
|
Balance at the beginning of the period |
|
$ |
|
|
$ |
- |
Investments |
|
|
|
|
|
|
Loss attributable to equity method investments |
|
|
( |
|
|
( |
Carrying value of equity method investments |
|
|
|
|
|
|
|
|
|
|
|
|
|
Non-marketable securities |
|
|
|
|
|
|
Balance at the beginning of the period |
|
$ |
|
|
$ |
|
Investments |
|
|
|
|
|
|
Impairment of investments |
|
|
( |
|
|
( |
Acquisition of controlling interest in previously held investment |
|
|
- |
|
|
( |
Carrying value of non-marketable securities |
|
|
|
|
|
|
Total investments in unconsolidated entities |
|
$ |
|
|
$ |
|
Accrued expenses at June 30, 2020 and June 30, 2019 consist of the following (in thousands):
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
||
Product warranties (note 8) |
|
$ |
|
|
$ |
|
Consulting and professional fees |
|
|
|
|
|
|
Value added taxes and other taxes due |
|
|
|
|
|
|
Employee related costs |
|
|
|
|
|
|
Hedging instruments |
|
|
|
|
|
|
Liability on receivables sold with recourse (note 17) |
|
|
|
|
|
|
Accrued interest |
|
|
|
|
|
|
Logistics and occupancy costs |
|
|
|
|
|
|
Inventory in transit |
|
|
|
|
|
|
Business acquisition contingent consideration |
|
|
|
|
|
- |
Litigation settlement expenses (note 20) |
|
|
- |
|
|
|
Restructuring expenses (note 19) |
|
|
- |
|
|
|
Other |
|
|
|
|
|
|
|
|
$ |
|
|
$ |
|
We include the liability for warranty costs in accrued expenses in our consolidated balance sheets. Changes in the liability for product warranty for the years ended June 30, 2020 and June 30, 2019 are as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
||
Balance at the beginning of the period |
|
$ |
|
|
$ |
|
Warranty accruals for the period |
|
|
|
|
|
|
Warranty costs incurred for the period |
|
|
( |
|
|
( |
Foreign currency translation adjustments |
|
|
( |
|
|
( |
Balance at the end of the period |
|
$ |
|
|
$ |
|
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
Debt at June 30, 2020 and June 30, 2019 consists of the following (in thousands):
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
||
Short-term debt |
|
$ |
|
|
$ |
|
Deferred borrowing costs |
|
|
( |
|
|
( |
Short-term debt, net |
|
|
|
|
|
|
|
|
|
- |
|
|
|
Long-term debt |
|
$ |
|
|
$ |
|
Deferred borrowing costs |
|
|
( |
|
|
( |
Long-term debt, net |
|
$ |
|
|
$ |
|
Total debt |
|
$ |
|
|
$ |
|
Credit Facility
On April 17, 2018, we entered into an amended and restated credit agreement (the “Revolving Credit Agreement”), as borrower, with lenders MUFG Union Bank, N.A., as administrative agent, joint lead arranger, joint book runner, swing line lender and letter of credit issuer, and Westpac Banking Corporation, as syndication agent, joint lead arranger and joint book runner. The Revolving Credit Agreement, among other things, provided a senior unsecured revolving credit facility of $
Additionally, on April 17, 2018, ResMed Limited entered into a Syndicated Facility Agreement (the “Term Credit Agreement”), as borrower, with lenders MUFG Union Bank, N.A., as administrative agent, joint lead arranger and joint book runner, and Westpac Banking Corporation, as syndication agent, joint lead arranger and joint book runner. The Term Credit Agreement, among other things, provides ResMed Limited a senior unsecured term credit facility of $
On November 5, 2018, we entered into a first amendment to the Revolving Credit Agreement to, among other things, increase the size of our senior unsecured revolving credit facility from $
Our obligations under the Revolving Credit Agreement are guaranteed by certain of our direct and indirect U.S. subsidiaries, and ResMed Limited’s obligations under the Term Credit Agreement are guaranteed by us and certain of our direct and indirect U.S. subsidiaries. The Revolving Credit Agreement and Term Credit Agreement contain customary covenants, including, in each case, a financial covenant that requires that we maintain a maximum leverage ratio of funded debt to EBITDA (as defined in the Revolving Credit Agreement and Term Credit Agreement, as applicable). The entire principal amounts of the revolving credit facility and term credit facility, and, in each case, any accrued but unpaid interest may be declared immediately due and payable if an event of default occurs, as defined in the Revolving Credit Agreement and the Term Credit Agreement, as applicable. Events of default under the Revolving Credit Agreement and the Term Credit Agreement include, in each case, failure to make payments when due, the occurrence of a default in the performance of any covenants in the respective agreements or related documents, or certain changes of control of us, or the respective guarantors of the obligations borrowed under the Revolving Credit Agreement and Term Credit Agreement.
The Revolving Credit Agreement and Term Credit Agreement each terminate on April 17, 2023, when all unpaid principal and interest under the loans must be repaid. Amounts borrowed under the Term Credit Agreement will also amortize on a semi-annual basis, with a $
We are required to disclose the fair value of financial instruments for which it is practicable to estimate the value, even though these instruments are not recognized at fair value in the consolidated balance sheets. As the Revolving Credit and Term Credit Agreements’ interest rate is calculated as LIBOR plus the spreads described above, its carrying amount is equivalent to its fair value as at June 30, 2020 and June 30, 2019, which was $
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
Senior Notes
On July 10, 2019, we entered into a Note Purchase Agreement with the purchasers to that agreement, in connection with the issuance and sale of $
Under the terms of the Note Purchase Agreement, we agreed to customary covenants including with respect to our corporate existence, transactions with affiliates, and mergers and other extraordinary transactions. We also agreed that, subject to limited exceptions, we will maintain a ratio of consolidated funded debt to consolidated EBITDA of no more than
We are required to disclose the fair value of financial instruments for which it is practicable to estimate the value, even though these instruments are not recognized at fair value in the consolidated balance sheets. As of June 30, 2020, the Senior Notes have a carrying amount of $
At June 30, 2020, we were in compliance with our debt covenants and there was $
(a) Leases where ResMed is the Lessee
We determine whether a contract is, or contains, a lease at inception. ROU assets represent our right to use an underlying asset during the lease term, and lease liabilities represent our obligation to make lease payments arising from the lease. ROU assets and lease liabilities are recognized at lease commencement based upon the estimated present value of unpaid lease payments over the lease term. We use our incremental borrowing rate based on the information available at lease commencement in determining the present value of unpaid lease payments. ROU assets also include any lease payments made at or before lease commencement and any initial direct costs incurred, and exclude any lease incentives received.
We determine the lease term as the non-cancellable period of the lease, and may include options to extend or terminate the lease when it is reasonably certain that we will exercise that option. Leases with a term of 12 months or less are not recognized on the balance sheet. Some of our leases include variable lease payments that are based on costs incurred or actual usage, or adjusted periodically based on an index or a rate. Our leases do not contain any residual value guarantees and we do not account for lease and non-lease components as a single lease component. Operating leases are included in operating lease right-of-use assets and operating lease liabilities on our consolidated balance sheets. We lease certain office space, warehouses and distribution centers, manufacturing facilities, vehicles, and equipment with remaining lease terms ranging from less than
Operating lease costs for the year ended June 30, 2020 were $
Future minimum lease payments under non-cancellable leases as of June 30, 2020 and for the periods ending June 30 of the years indicated below were as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
2021 |
|
2022 |
|
2023 |
|
2024 |
|
2025 |
|
Thereafter |
|||||||
Minimum lease payments |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
Less: imputed interest |
|
|
( |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total lease liabilities |
|
$ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
As of June 30, 2020, we had additional operating lease commitments of $
The supplemental information related to operating leases for the year ended June 30, 2020 was as follows (in thousands):
|
|
|
|
|
|
|
2020 |
||
Weighted-average inputs: |
|
|
|
|
Weighted-average remaining lease term (years) |
|
|
|
|
Weighted-average discount rate |
|
|
|
% |
|
|
|
|
|
Cash flow information: |
|
|
|
|
Operating cash flows paid for amounts included in the measurement of lease liabilities |
|
$ |
|
|
Right of use assets obtained in exchange for new lease liabilities: |
|
$ |
|
|
Disclosures related to periods prior to adopting the new lease guidance
We lease certain facilities and equipment under operating leases expiring at various dates and most contain renewal options. Total expense for all operating leases was $
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
2020 |
|
2021 |
|
2022 |
|
2023 |
|
2024 |
|
Thereafter |
|||||||
Remaining operating lease payments |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
(b) Leases where ResMed is the Lessor
We lease sleep and respiratory medical devices to customers primarily as a means to comply with local health insurer requirements in certain foreign geographies. Device rental contracts include sales-type and operating leases, and contract terms vary by customer and include options to terminate or extend the contract. When lease contracts also include the sale of masks and accessories, we allocate contract consideration to those items on a relative standalone price basis and recognize revenue when control transfers to the customer.
The components of lease revenue for the year ended June 30, 2020 were as follows (in thousands):
|
|
|
|
|
|
2020 |
|
Sales-type lease revenue |
|
$ |
|
Operating lease revenue |
|
|
|
Total lease revenue |
|
$ |
|
Our net investment in sales-type leases were classified in the accompanying consolidated balance sheets captions as of June 30, 2020 as follows (in thousands):
|
|
|
|
|
|
2020 |
|
Accounts receivable, net |
|
$ |
|
Prepaid taxes and other non-current assets |
|
|
|
Total |
|
$ |
|
Maturities of sales-type leases as of June 30, 2020 were as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Total |
|
2021 |
|
2022 |
|
2023 |
|
2024 |
|
2025 |
|
Thereafter |
|||||||
Remaining lease payments |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
- |
|
$ |
- |
|
$ |
- |
Less: imputed interest |
|
|
( |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Present value of remaining lease payments |
|
$ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
Common Stock. On February 21, 2014, our board of directors approved a new share repurchase program, authorizing us to acquire up to an aggregate of
We have temporarily suspended our repurchase program and, accordingly, did not repurchase any shares during fiscal year 2020. During fiscal year 2019, we repurchased
Preferred Stock. In April 1997, our board of directors authorized
Stock Options and Restricted Stock Units. We have granted stock options, restricted stock units (“RSUs”) and performance restricted stock units (“PRSUs”) to personnel, including officers and directors, in accordance with the ResMed Inc. 2009 Incentive Award Plan (the “2009 Plan”). Options and restricted stock units vest over
At the annual meeting of our stockholders in November 2017, our stockholders approved an amendment and restatement to the 2009 Plan to increase the number of shares of common stock that may be issued or transferred pursuant to awards under the 2009 Plan by
The maximum number of shares of our common stock authorized for issuance under the 2009 Plan is
In certain regions, shares are withheld on behalf of employees to satisfy statutory tax withholding requirements upon exercise or vesting of awards. The number of shares withheld is based upon the closing price of our common stock on the trading day of the applicable settlement date. The remaining shares are delivered to the recipient as shares of our common stock. The amount remitted to the tax authorities for the employees’ tax obligation is reflected as a financing activity on our consolidated statements of cash flows. Shares withheld by us as a result of the net settlement are not considered issued and outstanding and are added to the reserves of the 2009 Plan.
The total fair value of RSUs and PRSUs that vested during the years ended June 30, 2020, 2019 and 2018, was $
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
The following table summarizes the activity of RSUs, including PRSUs, during year ended June 30, 2020 (in thousands, except years and per share amounts):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Restricted |
|
Weighted |
|
Weighted |
||
Outstanding at beginning of period |
|
|
|
|
$ |
|
|
|
Granted |
|
|
|
|
|
|
|
|
Vested* |
|
|
( |
|
|
|
|
|
Performance factor adjustment |
|
|
|
|
|
|
|
|
Expired / cancelled |
|
|
( |
|
|
|
|
|
Forfeited |
|
|
( |
|
|
|
|
|
Outstanding at end of period |
|
|
|
|
$ |
|
|
|
*
The following table summarizes option activity during the year ended June 30, 2020 (in thousands, except years and per share amounts):
|
|
|
|
|
|
|
|
|
|
|
Options |
|
Weighted |
|
Weighted |
||
Outstanding at beginning of period |
|
|
|
|
$ |
|
|
|
Granted |
|
|
|
|
|
|
|
|
Exercised |
|
|
( |
|
|
|
|
|
Forfeited |
|
|
( |
|
|
|
|
|
Outstanding at end of period |
|
|
|
|
$ |
|
|
|
Options exercisable at end of period |
|
|
|
|
$ |
|
|
|
Options vested and expected to vest at end of period |
|
|
|
|
$ |
|
|
|
The aggregate intrinsic value of options exercised during the fiscal years 2020, 2019 and 2018, was $
Employee Stock Purchase Plan (the “ESPP”). Under the ESPP, we offer participants the right to purchase shares of our common stock at a discount during successive offering periods. Each offering period under the ESPP will be for a period of time determined by the board of directors’ compensation committee of no less than
During years ended June 30, 2020, 2019 and 2018, we issued
Stock–based Employee compensation. We measure the compensation expense of all stock-based awards at fair value on the grant date. We estimate the fair value of stock options and purchase rights granted under the ESPP using the Black-Scholes valuation model. The fair value of restricted stock units is equal to the market value of the underlying shares as determined at the grant date less the fair value of dividends that holders are not entitled to, during the vesting period. The fair value of performance restricted stock units is measured using a Monte-Carlo simulation valuation model. We recognize the fair value as compensation expense using the straight-line method over the service period for awards expected to vest.
We estimate the fair value of stock options granted under our stock option plans and purchase rights granted under the ESPP using the assumptions in the following tables. The risk-free interest rate is estimated using the U.S. Treasury yield curve and is based on the term of the award. The expected term of awards is estimated from the vesting period of the award, as well as historical exercise behavior, and represents the period of time the awards granted are expected to be outstanding. Expected volatility is estimated based upon the historical volatility of ResMed stock.
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
We estimate the fair value of stock options granted under our stock option plans and purchase rights granted under the ESPP using the
following assumptions for the years ended June 30, 2020, 2019 and 2018:
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
|
2018 |
|||
Stock options: |
|
|
|
|
|
|
|
|
|
Weighted average grant date fair value |
|
$ |
|
$ |
|
$ |
|||
Weighted average risk-free interest rate |
|
|
|
|
|
|
|||
Expected life in years |
|
|
|
|
|
|
|||
Dividend yield |
|
|
|
|
|
|
|||
Expected volatility |
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
ESPP purchase rights: |
|
|
|
|
|
|
|
|
|
Weighted average grant date fair value |
|
$ |
|
$ |
|
$ |
|||
Weighted average risk-free interest rate |
|
|
|
|
|
|
|||
Expected life in years |
|
|
|
|
|
|
|||
Dividend yield |
|
|
|
|
|
|
|||
Expected volatility |
|
|
|
|
|
|
|||
The following table summarizes the total stock-based compensation costs incurred and the associated tax benefit recognized during the years ended June 30, 2020, 2019 and 2018 (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
|
2018 |
|||
Cost of sales - capitalized as part of inventory |
|
$ |
|
|
$ |
|
|
$ |
|
Selling, general and administrative expenses |
|
|
|
|
|
|
|
|
|
Research and development expenses |
|
|
|
|
|
|
|
|
|
Stock-based compensation costs |
|
|
|
|
|
|
|
|
|
Tax benefit |
|
|
( |
|
|
( |
|
|
( |
Stock-based compensation costs, net of tax benefit |
|
$ |
|
|
$ |
|
|
$ |
|
At June 30, 2020, there was $
We compute basic earnings per share by dividing the net income available to common stockholders by the weighted average number of shares of common stock outstanding. For purposes of calculating diluted earnings per share, the denominator includes both the weighted average number of shares of common stock outstanding and the number of dilutive common stock equivalents such as stock options and restricted stock units. The weighted average number of outstanding stock options and restricted stock units not included in the computation of diluted earnings per share were
Basic and diluted earnings per share for the years ended June 30, 2020, 2019 and 2018 are calculated as follows (in thousands except per share data):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
|
2018 |
|||
Numerator: |
|
|
|
|
|
|
|
|
|
Net income |
|
$ |
|
|
$ |
|
|
$ |
|
Denominator: |
|
|
|
|
|
|
|
|
|
Basic weighted-average common shares outstanding |
|
|
|
|
|
|
|
|
|
Effect of dilutive securities: |
|
|
|
|
|
|
|
|
|
Stock options and restricted stock units |
|
|
|
|
|
|
|
|
|
Diluted weighted average shares |
|
|
|
|
|
|
|
|
|
Basic earnings per share |
|
$ |
|
|
$ |
|
|
$ |
|
Diluted earnings per share |
|
$ |
|
|
$ |
|
|
$ |
|
Other, net, in the consolidated statements of income is comprised of the following for the years ended June 30, 2020, 2019 and 2018 (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
|
2018 |
|||
Gain (loss) on foreign currency transactions and hedging, net |
|
$ |
|
|
$ |
|
|
$ |
( |
Impairment of equity investments (note 6) |
|
|
( |
|
|
( |
|
|
( |
Other |
|
|
|
|
|
|
|
|
|
|
|
$ |
( |
|
$ |
( |
|
$ |
( |
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
Income before income taxes for the years ended June 30, 2020, 2019 and 2018, was taxed under the following jurisdictions (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
|
2018 |
|||
U.S. |
|
$ |
|
|
$ |
( |
|
$ |
|
Non-U.S. |
|
|
|
|
|
|
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
The provision for income taxes is presented below (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
|
2018 |
|||
Current: |
Federal |
|
$ |
|
|
$ |
|
|
$ |
|
|
State |
|
|
|
|
|
|
|
|
|
|
Non-U.S. |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deferred: |
Federal |
|
|
( |
|
|
( |
|
|
|
|
State |
|
|
( |
|
|
( |
|
|
( |
|
Non-U.S. |
|
|
( |
|
|
( |
|
|
( |
|
|
|
|
( |
|
|
( |
|
|
|
Provision for income taxes |
|
$ |
|
|
$ |
|
|
$ |
|
|
The provision for income taxes differs from the amount of income tax determined by applying the applicable U.S. federal income tax rate of
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
|
2018 |
|||
Taxes computed at statutory U.S. rate |
|
$ |
|
|
$ |
|
|
$ |
|
Increase (decrease) in income taxes resulting from: |
|
|
|
|
|
|
|
|
|
Transition tax |
|
|
- |
|
|
|
|
|
|
State income taxes, net of U.S. tax benefit |
|
|
|
|
|
|
|
|
|
Research and development credit |
|
|
( |
|
|
( |
|
|
( |
Change in statutory tax rates |
|
|
- |
|
|
- |
|
|
|
Change in valuation allowance |
|
|
|
|
|
( |
|
|
( |
Effect of non-U.S. tax rates |
|
|
( |
|
|
|
|
|
( |
Foreign tax credits (1) |
|
|
( |
|
|
( |
|
|
( |
Stock-based compensation expense |
|
|
( |
|
|
( |
|
|
( |
Other |
|
|
|
|
|
|
|
|
|
|
|
$ |
|
|
$ |
|
|
$ |
|
(1)
The components of our deferred tax assets and liabilities at June 30, 2020 and June 30, 2019, are as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
||
Deferred tax assets: |
|
|
|
|
|
|
|
|
|
Employee liabilities |
|
|
|
|
$ |
|
|
$ |
|
Tax credit carry overs |
|
|
|
|
|
|
|
|
|
Inventories |
|
|
|
|
|
|
|
|
|
Provision for warranties |
|
|
|
|
|
|
|
|
|
Provision for doubtful debts |
|
|
|
|
|
|
|
|
|
Net operating loss carryforwards |
|
|
|
|
|
|
|
|
|
Capital loss carryover |
|
|
|
|
|
|
|
|
|
Property, plant and equipment |
|
|
|
|
|
( |
|
|
|
Stock-based compensation expense |
|
|
|
|
|
|
|
|
|
Deferred revenue |
|
|
|
|
|
|
|
|
|
Research and development capitalization |
|
|
|
|
|
|
|
|
|
Other |
|
|
|
|
|
( |
|
|
( |
|
|
|
|
|
|
|
|
|
|
Less valuation allowance |
|
|
|
|
|
( |
|
|
( |
Deferred tax assets |
|
|
|
|
|
|
|
|
|
Deferred tax liabilities: |
|
|
|
|
|
|
|
|
|
Goodwill and other intangibles |
|
|
|
|
|
( |
|
|
( |
Deferred tax liabilities |
|
|
|
|
|
( |
|
|
( |
Net deferred tax asset |
|
|
|
|
$ |
|
|
$ |
|
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
We reported the net deferred tax assets and liabilities in our consolidated balance sheets at June 30, 2020 and June 30, 2019, as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
||
Non-current deferred tax asset |
|
|
|
|
$ |
|
|
$ |
|
Non-current deferred tax liability |
|
|
|
|
|
( |
|
|
( |
Net deferred tax asset |
|
|
|
|
$ |
|
|
$ |
|
As of June 30, 2020, we had $
The valuation allowance at June 30, 2020 relates to a provision for uncertainty of the utilization of net operating loss carryforwards of $
A substantial portion of our manufacturing operations and administrative functions in Singapore operate under various tax holidays and tax incentive programs that will expire in whole or in part at various dates through June 30, 2030. The end of certain tax holidays may be extended if specific conditions are met. The net impact of these tax holidays and tax incentive programs increased our net earnings by $
As a result of the U.S. Tax Act, we have treated all non-U.S. historical earnings as taxable, which resulted in additional tax expense of $
In accounting for uncertainty in income taxes, we recognize a tax benefit in the financial statements for an uncertain tax position only if management’s assessment is that the position is “more likely than not” (that is, a likelihood greater than
Our income tax returns are based on calculations and assumptions subject to audit by various tax authorities. In addition, the calculation of our tax liabilities involves dealing with uncertainties in the application of complex tax laws. We regularly assess the potential outcomes of examinations by tax authorities in determining the adequacy of our provision for income taxes. Any final assessment resulting from tax audits may result in material changes to our past or future taxable income, tax payable or deferred tax assets, and may require us to pay penalties and interest that could materially adversely affect our financial results.
In connection with the audit by the Australian Taxation Office (“ATO”) for the tax years
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
Our income tax expense, short-term income taxes payable and long-term income taxes payable were impacted by charges associated with the U.S. Tax Act enacted on December 22, 2017, which resulted in additional income tax expense of $
We have
Certain items are maintained at the corporate level and are not allocated to the segments. The non-allocated items include corporate headquarters costs, stock-based compensation, amortization expense from acquired intangibles, acquisition related expenses, net interest expense and other, net. We neither discretely allocate assets to our operating segments, nor does our Chief Operating Decision Maker evaluate the operating segments using discrete asset information.
The table below presents a reconciliation of net revenues, depreciation and amortization and net operating profit by reportable segments for the years ended June 30, 2020, 2019 and 2018 (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
|
2018 |
|||
Revenue by segment |
|
|
|
|
|
|
|
|
|
Total Sleep and Respiratory Care |
|
$ |
|
|
$ |
|
|
$ |
|
|
|
|
|
|
|
|
|
|
|
Software as a Service |
|
|
|
|
|
|
|
|
|
Deferred revenue fair value adjustment* |
|
|
( |
|
|
( |
|
|
- |
Total Software as a Service |
|
|
|
|
|
|
|
|
|
Total |
|
$ |
|
|
$ |
|
|
$ |
|
|
|
|
|
|
|
|
|
|
|
Depreciation and amortization by segment |
|
|
|
|
|
|
|
|
|
Sleep and Respiratory Care |
|
$ |
|
|
$ |
|
|
$ |
|
Software as a Service |
|
|
|
|
|
|
|
|
|
Amortization of acquired intangible assets and corporate costs |
|
|
|
|
|
|
|
|
|
Total |
|
$ |
|
|
$ |
|
|
$ |
|
|
|
|
|
|
|
|
|
|
|
Net operating profit by segment |
|
|
|
|
|
|
|
|
|
Sleep and Respiratory Care |
|
$ |
|
|
$ |
|
|
$ |
|
Software as a Service |
|
|
|
|
|
|
|
|
|
Total |
|
$ |
|
|
$ |
|
|
$ |
|
|
|
|
|
|
|
|
|
|
|
Reconciling items |
|
|
|
|
|
|
|
|
|
Corporate costs |
|
$ |
|
|
$ |
|
|
$ |
|
Amortization of acquired intangible assets |
|
|
|
|
|
|
|
|
|
Litigation settlement expenses |
|
|
( |
|
|
|
|
|
- |
Restructuring expenses |
|
|
- |
|
|
|
|
|
|
Acquisition related expenses |
|
|
- |
|
|
|
|
|
- |
Deferred revenue fair value adjustment* |
|
|
|
|
|
|
|
|
- |
Interest expense (income), net |
|
|
|
|
|
|
|
|
|
Loss attributable to equity method investments |
|
|
|
|
|
|
|
|
- |
Other, net |
|
|
|
|
|
|
|
|
|
Income before income taxes |
|
$ |
|
|
$ |
|
|
$ |
|
*
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
The following table summarizes our net revenue disaggregated by segment, product and region for the years ended June 30, 2020, 2019 and 2018 (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
|
2018 |
|||
U.S., Canada and Latin America |
|
|
|
|
|
|
|
|
|
Devices |
|
$ |
|
|
$ |
|
|
$ |
|
Masks and other |
|
|
|
|
|
|
|
|
|
Total Sleep and Respiratory Care |
|
$ |
|
|
$ |
|
|
$ |
|
Software as a Service |
|
|
|
|
|
|
|
|
|
Total |
|
$ |
|
|
$ |
|
|
$ |
|
|
|
|
|
|
|
|
|
|
|
Combined Europe, Asia and other markets |
|
|
|
|
|
|
|
|
|
Devices |
|
$ |
|
|
$ |
|
|
$ |
|
Masks and other |
|
|
|
|
|
|
|
|
|
Total Sleep and Respiratory Care |
|
$ |
|
|
$ |
|
|
$ |
|
|
|
|
|
|
|
|
|
|
|
Global revenue |
|
|
|
|
|
|
|
|
|
Devices |
|
$ |
|
|
$ |
|
|
$ |
|
Masks and other |
|
|
|
|
|
|
|
|
|
Total Sleep and Respiratory Care |
|
$ |
|
|
$ |
|
|
$ |
|
Software as a Service |
|
|
|
|
|
|
|
|
|
Total |
|
$ |
|
|
$ |
|
|
$ |
|
Revenue information by geographic area for the years ended June 30, 2020, 2019 and 2018 is summarized below (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
|
2018 |
|||
United States |
|
$ |
|
|
$ |
|
|
$ |
|
Rest of the World |
|
|
|
|
|
|
|
|
|
Total |
|
$ |
|
|
$ |
|
|
$ |
|
Long-lived assets of geographic areas are those assets used in our operations in each geographical area, and excludes goodwill, other intangible assets, and deferred tax assets. Long-lived assets by geographic area as of June 30, 2020, 2019 and 2018, is summarized below (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
|
2018 |
|||
United States |
|
$ |
|
|
$ |
|
|
$ |
|
Australia |
|
|
|
|
|
|
|
|
|
Singapore |
|
|
|
|
|
|
|
|
|
Rest of the World |
|
|
|
|
|
|
|
|
|
Total |
|
$ |
|
|
$ |
|
|
$ |
|
We contribute to a number of employee retirement plans for the benefit of our employees. Details of the main plans are as follows:
Australia We contribute to defined contribution plans for each employee resident in Australia at the rate of approximately
United States We sponsor a defined contribution plan available to substantially all domestic employees. Company contributions to this plan are based on a percentage of employee contributions to a maximum of
Singapore We sponsor a defined contribution plan available to substantially all domestic employees. Company contributions to this plan are based on a percentage of employee contributions to a maximum of
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
Litigation
In the normal course of business, we are subject to routine litigation incidental to our business. While the results of this litigation cannot be predicted with certainty, we believe that their final outcome will not, individually or in aggregate, have a material adverse effect on our consolidated financial statements taken as a whole.
Taxation Matters
As described in note 14 – Income Taxes, we received Notices of Amended Assessments from the ATO for the tax years 2009 to 2013. Based on these assessments, the ATO asserted that we owe $
Contingent Obligations Under Recourse Provisions
We use independent financing institutions to offer some of our customers financing for the purchase of some of our products. Under these arrangements, if the customer qualifies under the financing institutions’ credit criteria and finances the transaction, the customers repay the financing institution on a fixed payment plan. For some of these arrangements, the customer’s receivable balance is with recourse, either limited or full, whereby we are responsible for repaying the financing company should the customer default. We record a contingent provision, which is estimated based on historical default rates. This is applied to receivables sold with recourse and is recorded in accrued expenses.
The following table summarizes the amount of total receivables sold with recourse during the years ended June 30, 2020 and June 30, 2019 (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
||
Full recourse |
|
$ |
- |
|
$ |
|
Limited recourse |
|
|
|
|
|
|
Total |
|
$ |
|
|
$ |
|
The following table summarizes the maximum exposure on outstanding receivables sold with recourse and provision for doubtful accounts as at June 30, 2020 and June 30, 2019 (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
2020 |
|
2019 |
||
Full recourse |
|
$ |
|
|
$ |
|
Limited recourse |
|
|
|
|
|
|
Total |
|
$ |
|
|
$ |
|
Contingent provision for receivables with recourse |
|
$ |
( |
|
$ |
( |
Commitments
In the normal course of business, we enter into agreements to purchase goods or services that are not cancelable without penalty, primarily related to supply arrangements. Obligations under our purchase agreements at June 30, 2020 were as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Fiscal Years Ending June 30 |
||||||||||||||||
|
|
Total |
|
2021 |
|
2022 |
|
2023 |
|
2024 |
|
2025 |
|
Thereafter |
|||||||
Minimum purchase obligations |
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
|
|
$ |
- |
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
Fiscal year ended June 30, 2020
On January 31, 2020, we completed the acquisition of
We completed our purchase price allocation during the quarter ending June 30, 2020, which was not materially different from the preliminary purchase price allocation. The cost of the acquisition was allocated to the assets acquired and liabilities assumed based on estimates of their fair values at the date of acquisition. The goodwill recognized as part of the acquisition is reflected in the Software as a Service segment and is deductible for tax purposes. It mainly represents the synergies that are unique to our combined businesses and the potential for new products and services to be developed in the future.
During the year ended June 30, 2020 we did
Fiscal year ended June 30, 2019
MatrixCare
On November 13, 2018, we completed the acquisition of
We completed the purchase price allocation in relation to this acquisition during the quarter ended December 31, 2019. The cost of the acquisition was allocated to the assets acquired and liabilities assumed based on estimates of their fair values at the date of acquisition. The goodwill recognized as part of the acquisition is reflected in the Software as a Service segment and is not deductible for tax purposes. It mainly represents the synergies that are unique to our combined businesses and the potential for new products and services to be developed in the future. The fair values of assets acquired and liabilities assumed, and the estimated useful lives of intangible assets acquired are as follows (in thousands):
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Preliminary as of |
|
Adjustments |
|
Final |
|
Intangible |
||||
Current assets |
|
$ |
|
|
$ |
- |
|
$ |
|
|
|
|
Property, plant and equipment |
|
|
|
|
|
- |
|
|
|
|
|
|
Trade names |
|
|
|
|
|
- |
|
|
|
|
|
|
Developed technology |
|
|
|
|
|
- |
|
|
|
|
|
|
Customer relationships |
|
|
|
|
|
|
|
|
|
|
|
|
Goodwill |
|
|
|
|
|
|
|
|
|
|
|
|
Assets acquired |
|
$ |
|
|
$ |
|
|
$ |
|
|
|
|
Current liabilities |
|
|
( |
|
|
( |
|
|
( |
|
|
|
Deferred revenue |
|
|
( |
|
|
( |
|
|
( |
|
|
|
Deferred tax liabilities |
|
|
( |
|
|
( |
|
|
( |
|
|
|
Debt assumed |
|
|
( |
|
|
- |
|
|
( |
|
|
|
Total liabilities assumed |
|
$ |
( |
|
$ |
( |
|
$ |
( |
|
|
|
Net assets acquired |
|
$ |
|
|
$ |
- |
|
$ |
|
|
|
|
A reconciliation of the base consideration to the net consideration is as follows (in thousands):
|
|
|
|
Base consideration |
|
$ |
|
Cash acquired |
|
|
|
Debt assumed |
|
|
( |
Net working capital and other adjustments |
|
|
( |
Net consideration |
|
$ |
|
During the year ended June 30, 2019, revenues of $
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
deferred revenue of $
The acquisition is considered a material business combination and accordingly unaudited pro forma information presented below for the year ended June 30, 2019, includes the effects of pro forma adjustments as if the acquisition of MatrixCare occurred on July 1, 2017. The pro forma results were prepared using the acquisition method of accounting and combine our historical results and MatrixCare’s for the years ended June 30, 2019 and June 30, 2018, including the effects of the business combination, primarily amortization expense related to the fair value of identifiable intangible assets acquired, interest expense associated with the financing obtained by us in connection with the acquisition, and the elimination of incurred acquisition-related costs.
The pro forma financial information presented below is not necessarily indicative of the results of operations that would have been achieved if the acquisition occurred at the beginning of the earliest period presented, nor is it intended to be a projection of future results. The following table summarized unaudited pro forma consolidated results for the years ended June 30, 2019 and 2018 (in thousands, except per share information):
|
|
|
|
|
|
|
2019 |
|
2018 |
||
Revenue |
$ |
|
|
$ |
|
Net income |
$ |
|
|
$ |
|
Basic earnings per share |
$ |
|
|
$ |
|
Diluted earnings per share |
$ |
|
|
$ |
|
The unaudited pro forma consolidated results for the years ended June 30, 2019 and June 30, 2018 reflect primarily the following pro forma pre-tax adjustments:
Net amortization expense related to the fair value of identifiable intangible assets acquired of $
Net interest expense associated with debt that was issued to finance the acquisition of $
Elimination of pre-tax acquisition-related costs incurred by ResMed and MatrixCare of $
Net income tax expense of $
Other acquisitions
During the year ended June 30, 2019, we also completed the following acquisitions:
On July 6, 2018, we completed the acquisition of
On October 15, 2018, we completed the acquisition of
On December 11, 2018, we completed the acquisition of assets in Interactive Health Network, a provider of integrated clinical and financial management software solution for long-term care companies.
On December 13, 2018, we completed the acquisition of assets in Apacheta, a provider of cloud-based SaaS software that manages the medical equipment delivery process for HME dealers.
On January 6, 2019, we completed the acquisition of Propeller Health, a digital therapeutics company providing connected health solutions for people living with chronic obstructive pulmonary disease and asthma, for a total purchase consideration of $
These acquisitions have been accounted for as business combinations using purchase accounting and are included in our consolidated financial statements from the acquisition dates. These acquisitions, individually and collectively, are not considered a material business combination and accordingly pro forma information is not provided. The acquisitions were funded by drawing on our existing revolving credit facility and through cash on-hand.
Table of Contents
PART II |
Item 8 |
RESMED INC. AND SUBSIDIARIES
Notes to the Consolidated Financial Statements
We have completed the purchase price allocation in relation to all of these acquisitions. The cost of the share acquisitions was allocated to the assets acquired and liabilities assumed based on estimates of their fair values at the date of acquisition. The goodwill recognized as part of these acquisitions, which is predominantly not deductible for tax purposes, mainly represents the synergies that are unique to our combined businesses and the potential for new products and services to be developed in the future. Goodwill from these acquisitions has been reflected in the Software as a Service segment except for the goodwill resulting from the HB Healthcare and Propeller Health acquisitions, which have been recorded in the Sleep and Respiratory Care segment.
The fair values of assets acquired and liabilities assumed of acquisitions during the year ended June 30, 2019, excluding MatrixCare, and the estimated useful lives of intangible assets acquired are as follows (in thousands):
|
|
|
|
|
|
|
|
|
Final |
|
Intangible |
||
Current assets |
|
$ |
|
|
|
|
Property, plant and equipment |
|
|
|
|
|
|
Deferred tax assets |
|
|
|
|
|
|
Trade names |
|
|
|
|
|
|
Non-compete |
|
|
|
|
|
|
Developed technology |
|
|
|
|
|
|
Customer relationships |
|
|
|
|
|
|
Goodwill |
|
|
|
|
|
|
Assets acquired |
|
$ |
|
|
|
|
Current liabilities |
|
|
( |
|
|
|
Deferred revenue |
|
|
( |
|
|
|
Deferred tax liabilities |
|
|
( |
|
|
|
Debt assumed |
|
|
( |
|
|
|
Total liabilities assumed |
|
$ |
( |
|
|
|
Net assets acquired |
|
$ |
|
|
|
|
During the year ended June 30, 2020, we did not incur material restructuring expenses.
During the year ended June 30, 2019, we incurred restructuring expenses of $
During the year ended June 30, 2018, we incurred restructuring expenses within the Sleep and Respiratory Care segment of $
We did
RESMED INC. AND SUBSIDIARIES
VALUATION AND QUALIFYING ACCOUNTS AND RESERVES
June 30, 2020, 2019 and 2018
(in thousands)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Balance at |
|
Charged to costs and expenses |
|
Other |
|
Balance at |
||||
Year ended June 30, 2020 |
|
|
|
|
|
|
|
|
|
|
|
|
Applied against asset account |
|
|
|
|
|
|
|
|
|
|
|
|
Allowance for doubtful accounts |
|
$ |
|
|
$ |
|
|
$ |
( |
|
$ |
|
Year ended June 30, 2019 |
|
|
|
|
|
|
|
|
|
|
|
|
Applied against asset account |
|
|
|
|
|
|
|
|
|
|
|
|
Allowance for doubtful accounts |
|
$ |
|
|
$ |
|
|
$ |
( |
|
$ |
|
Year ended June 30, 2018 |
|
|
|
|
|
|
|
|
|
|
|
|
Applied against asset account |
|
|
|
|
|
|
|
|
|
|
|
|
Allowance for doubtful accounts |
|
$ |
|
|
$ |
|
|
$ |
( |
|
$ |
|
ITEM 9 CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A CONTROLS AND PROCEDURES
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our Exchange Act reports is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms and that such information is accumulated and communicated to our management, including our chief executive officer and chief financial officer, as appropriate, to allow for timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management is required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
As required by SEC Rule 13a-15(b), we carried out an evaluation, under the supervision and with the participation of our management, including our chief executive officer and chief financial officer, of the effectiveness of the design and operation of our disclosure controls and procedures as of June 30, 2020. Based on the foregoing, our chief executive officer and chief financial officer concluded that our disclosure controls and procedures were effective at the reasonable assurance level as of June 30, 2020.
There has been no change in our internal control over financial reporting during our most recent fiscal quarter that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting as defined in Rules 13a-15(f) and 15d-15(f) under the Securities Exchange Act of 1934, as amended. Our internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles in the United States of America. Our internal control over financial reporting includes those policies and procedures that:
(i)Pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets;
(ii)Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and
(iii)Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Management assessed the effectiveness of our internal control over financial reporting as of June 30, 2020. In making this assessment, management used the framework in Internal Control-Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. Management’s assessment included an evaluation of the design of our internal control over financial reporting and testing of the operational effectiveness of its internal control over financial reporting. Management reviewed the results of its assessment with the audit committee of our board of directors.
Based on that assessment under the framework in Internal Control-Integrated Framework (2013), management concluded that the company’s internal control over financial reporting was effective as of June 30, 2020.
KPMG LLP, independent registered public accounting firm, who audited and reported on the consolidated financial statements of ResMed, Inc. included in this report, has issued an attestation report on the effectiveness of internal control over financial reporting.
Report of Independent Registered Public Accounting Firm
To the Stockholders and Board of Directors
ResMed Inc.:
Opinion on Internal Control Over Financial Reporting
We have audited ResMed Inc. and subsidiaries’ (the Company) internal control over financial reporting as of June 30, 2020, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission. In our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of June 30, 2020, based on criteria established in Internal Control – Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States) (PCAOB), the consolidated balance sheets of the Company as of June 30, 2020 and 2019, the related consolidated statements of income, comprehensive income, stockholders’ equity, and cash flows for each of the years in the three-year period ended June 30, 2020, and the related notes and financial statement schedule II (collectively, the consolidated financial statements), and our report dated August 12, 2020 expressed an unqualified opinion on those consolidated financial statements.
Basis for Opinion
The Company’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We are a public accounting firm registered with the PCAOB and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audit in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audit also included performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
Definition and Limitations of Internal Control Over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
San Diego, California
August 12, 2020
ITEM 9B OTHER INFORMATION
None.
PART III
ITEM 10 DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Information required by this Item is incorporated by reference from our definitive proxy statement for our next annual meeting of stockholders, which will be filed with the Securities and Exchange Commission within 120 days after June 30, 2020.
We have filed as exhibits to this report for the year ended June 30, 2020, the certifications of our chief executive officer and chief financial officer required by Section 302 of the Sarbanes-Oxley Act of 2002.
ITEM 11 EXECUTIVE COMPENSATION
Information required by this Item is incorporated by reference from our definitive proxy statement for our next annual meeting of stockholders, which will be filed with the Securities and Exchange Commission within 120 days after June 30, 2020.
ITEM 12 SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
Information required by this Item is incorporated by reference from our definitive proxy statement for our next annual meeting of stockholders, which will be filed with the Securities and Exchange Commission within 120 days after June 30, 2020.
ITEM 13 CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
Information required by this Item is incorporated by reference from our definitive proxy statement for our next annual meeting of stockholders, which will be filed with the Securities and Exchange Commission within 120 days after June 30, 2020.
ITEM 14 PRINCIPAL ACCOUNTING FEES AND SERVICES
Information required by this Item is incorporated by reference from our definitive proxy statement for our next annual meeting of stockholders, which will be filed with the Securities and Exchange Commission within 120 days after June 30, 2020.
PART IV
ITEM 15 EXHIBITS AND CONSOLIDATED FINANCIAL STATEMENT SCHEDULES
The following documents are filed as part of this report:
|
|
|
|
(a) |
Consolidated Financial Statements and Schedules – The index to our consolidated financial statements and schedules are set forth in the “Index to Consolidated Financial Statements” under Item 8 of this report. |
|
|
(b) |
Exhibit Lists |
|
|
2.1 |
Agreement and Plan of Merger, dated November 5, 2018, by and among ResMed Operations Inc., Evolved Sub, Inc., ResMed Inc., OPEL GI Holdings Limited, in its capacity as the agent acting on behalf of the holders of common stock of MatrixCare Holdings, Inc., and MatrixCare Holdings, Inc. (Incorporated by reference to Exhibit 2.1 to the Registrant’s Report on Form 8-K filed on November 8, 2018) |
3.1 |
First Restated Certificate of Incorporation of ResMed Inc., as amended. (Incorporated by reference to Exhibit 3.1 to the Registrant’s Report on Form 10-Q for the quarter ended September 30, 2013) |
3.2 |
Sixth Amended and Restated Bylaws of ResMed Inc. (Incorporated by reference to Exhibit 3.1 to the Registrant’s Report on Form 8-K filed on February 26, 2020) |
4.1 |
Form of certificate evidencing shares of Common Stock. (Incorporated by reference to Exhibit 4.1 to the Registrant’s Registration Statement on Form S-1 (No. 33-91094) declared effective on June 1, 1995) |
4.2 |
|
10.1 |
Licensing Agreement between the University of Sydney and ResMed Ltd dated May 17, 1991, as amended. (Incorporated by reference to Exhibit 10.2 to the Registrant’s Registration Statement on Form S-1 (No. 33-91094) declared effective on June 1, 1995) |
10.2* |
ResMed Inc. 2006 Incentive Award Plan. (Incorporated by reference to Exhibit 99.1 to the Registrant’s Report on Form 8-K filed on November 15, 2006) |
10.3* |
Amendment No. 1 to the ResMed Inc. 2006 Incentive Award Plan. (Incorporated by reference to Exhibit 10.24 to the Registrant’s Report on Form 10-Q for the quarter ended December 31, 2006, filed on February 8, 2007) |
10.4* |
2006 Grant agreement for Board of Directors. (Incorporated by reference to Exhibit 10.25 to the Registrant’s Report on Form 10-Q for the quarter ended December 31, 2006 filed on February 8, 2007) |
10.5* |
2006 Grant agreement for Executive Officers. (Incorporated by reference to Exhibit 10.26 to the Registrant’s Report on Form 10-K for the year ended June 30, 2007, filed on August 28, 2007) |
10.6* |
2006 Grant agreement for Australian Executive Officers. (Incorporated by reference to Exhibit 10.27 to the Registrant’s Report on Form 10-K for the year ended June 30, 2007, filed on August 28, 2007) |
10.7* |
Form of Executive Agreement. (Incorporated by reference to Exhibit 99.1 to the Registrant’s Report on Form 8-K filed on July 13, 2007) |
10.8* |
Amended and Restated 2006 Incentive Award Plan dated November 20, 2008. (Incorporated by reference to Appendix 1 of the Registrant’s Definitive Proxy Statement filed on October 15, 2008) |
10.9 |
Form of Indemnification Agreements for our directors and officers. (Incorporated by reference to Exhibit 10.1 the Registrant’s Report on Form 8-K filed on June 24, 2009) |
10.10 |
Form of Access Agreement for directors. (Incorporated by reference to Exhibit 10.2 the Registrant’s Report on Form 8-K filed on June 24, 2009) |
10.11* |
Updated Form of Executive Agreement. (Incorporated by reference to Exhibit 99.1 to the Registrant’s Report on Form 8-K filed on July 2, 2012) |
10.12 |
Amendment and Restatement to the ResMed Inc. 2009 Incentive Award Plan. (Incorporated by reference to Appendix B of ResMed Inc.’s Proxy Statement filed with the Securities and Exchange Commission on September 25, 2017.) |
10.13 |
ResMed Inc. 2009 Incentive Award Plan. (Incorporated by reference to Exhibit 10.1 the Registrant’s Report on Form 8-K filed on November 23, 2009) |
10.14 |
ResMed Inc. Deferred Compensation Plan. (Incorporated by reference to Exhibit 10.1 the Registrant’s Report on Form 8-K filed on May 28, 2010) |
10.15 |
Form of Restricted Stock Unit Award Agreement for Executive Officers. (Incorporated by reference to Exhibit 10.1 to the Registrant’s Report on Form 10-Q for the quarter ended September 30, 2011, filed on November 3, 2011) |
10.16 |
Form of Restricted Stock Unit Award Agreement for Directors. (Incorporated by reference to Exhibit 10.2 to the Registrant’s Report on Form 10-Q for the quarter ended September 30, 2011, filed on November 3, 2011) |
10.17 |
Form of Stock Option Grant for Executive Officers. (Incorporated by reference to Exhibit 10.3 to the Registrant’s Report on Form 10-Q for the quarter ended September 30, 2011, filed on November 3, 2011) |
* Management contract or compensatory plan or arrangement
ITEM 16 FORM 10-K SUMMARY
None.
SIGNATURES
Under the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has caused this report to be signed on its behalf by the authorized persons below.
DATED August 12, 2020
ResMed Inc.
/s/ MICHAEL J. FARRELL |
Michael J. Farrell |
Chief executive officer |
(Principal Executive Officer) |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated.
SIGNATURE |
|
TITLE |
|
DATE |
|
|
|
|
|
/S/ MICHAEL J. FARRELL |
|
Chief executive officer and director |
|
August 12, 2020 |
Michael J. Farrell |
|
(Principal Executive Officer) |
|
|
|
|
|
|
|
/S/ BRETT A. SANDERCOCK |
|
Chief financial officer |
|
August 12, 2020 |
Brett A. Sandercock |
|
(Principal Financial Officer and Principal Accounting Officer) |
|
|
|
|
|
|
|
/S/ PETER C. FARRELL |
|
Non-executive chairman |
|
August 12, 2020 |
Peter C. Farrell |
|
|
|
|
|
|
|
|
|
/S/ CAROL J. BURT |
|
Director |
|
August 12, 2020 |
Carol J. Burt |
|
|
|
|
|
|
|
|
|
/S/ JAN De WITTE |
|
Director |
|
August 12, 2020 |
Jan De Witte |
|
|
|
|
|
|
|
|
|
/S/ RICHARD SULPIZIO |
|
Director |
|
August 12, 2020 |
Richard Sulpizio |
|
|
|
|
|
|
|
|
|
/S/ RON TAYLOR |
|
Director |
|
August 12, 2020 |
Ron Taylor |
|
|
|
|
|
|
|
|
|
/S/ KAREN DREXLER |
|
Director |
|
August 12, 2020 |
Karen Drexler |
|
|
|
|
|
|
|
|
|
/S/ HARJIT GILL |
|
Director |
|
August 12, 2020 |
Harjit Gill |
|
|
|
|
|
|
|
|
|